Functions from these packages will be used throughout this document:
[R code]
library(conflicted) # check for conflicting function definitions# library(printr) # inserts help-file output into markdown outputlibrary(rmarkdown) # Convert R Markdown documents into a variety of formats.library(pander) # format tables for markdownlibrary(ggplot2) # graphicslibrary(ggfortify) # help with graphicslibrary(dplyr) # manipulate datalibrary(tibble) # `tibble`s extend `data.frame`slibrary(magrittr) # `%>%` and other additional piping toolslibrary(haven) # import Stata fileslibrary(knitr) # format R output for markdownlibrary(tidyr) # Tools to help to create tidy datalibrary(plotly) # interactive graphicslibrary(dobson) # datasets from Dobson and Barnett 2018library(parameters) # format model output tables for markdownlibrary(haven) # import Stata fileslibrary(latex2exp) # use LaTeX in R code (for figures and tables)library(fs) # filesystem path manipulationslibrary(survival) # survival analysislibrary(survminer) # survival analysis graphicslibrary(KMsurv) # datasets from Klein and Moeschbergerlibrary(parameters) # format model output tables forlibrary(webshot2) # convert interactive content to static for pdflibrary(forcats) # functions for categorical variables ("factors")library(stringr) # functions for dealing with stringslibrary(lubridate) # functions for dealing with dates and times
Here are some R settings I use in this document:
[R code]
rm(list =ls()) # delete any data that's already loaded into Rconflicts_prefer(dplyr::filter)ggplot2::theme_set( ggplot2::theme_bw() +# ggplot2::labs(col = "") + ggplot2::theme(legend.position ="bottom",text = ggplot2::element_text(size =12, family ="serif")))knitr::opts_chunk$set(message =FALSE)options('digits'=6)panderOptions("big.mark", ",")pander::panderOptions("table.emphasize.rownames", FALSE)pander::panderOptions("table.split.table", Inf)conflicts_prefer(dplyr::filter) # use the `filter()` function from dplyr() by defaultlegend_text_size =9run_graphs =TRUE
1 Introduction
Exercise 1 Recall the key characteristics of the exponential distribution:
Definition 1 (conditional hazard) The conditional hazard of outcome \(T\) at value \(t\), given covariate vector \(\tilde{x}\), is the conditional density of the event \(T=t\), given \(T \ge t\) and \(\tilde{X}= \tilde{x}\):
Definition 4 (Baseline survival function) The baseline survival function is the survival function for an individual whose covariates are all equal to their default values.
Definition 8 (difference in log-hazards) The difference in log-hazards between covariate patterns \(\tilde{x}\) and \({\tilde{x}^*}\) at time \(t\) is:
Theorem 2 (Difference of log-hazards vs hazard ratio) If \(\Delta \eta(t|\tilde{x}: {\tilde{x}^*})\) is the difference in log-hazard between covariate patterns \(\tilde{x}\) and \({\tilde{x}^*}\) at time \(t\), and \(\theta(t| \tilde{x}: {\tilde{x}^*})\) is corresponding hazard ratio, then:
Corollary 1 (Hazard ratio vs difference of log-odds)\[\theta(t| \tilde{x}: {\tilde{x}^*}) = \text{exp}{\left\{\Delta \eta(t|\tilde{x}: {\tilde{x}^*})\right\}}\]
Definition 9 (difference in log-hazard from baseline)
Definition 11 (Proportional hazards model) A proportional hazards model for a time-to-event outcome \(T\) is a model where the difference in log-hazard from the baseline log-hazard is equal to a linear combination of the predictors:
Definition 12 (proportional hazards) A conditional probability distribution \(p(T|X)\) has proportional hazards if the hazard ratio \({\lambda}(t|\tilde{x}_1)/{\lambda}(t|\tilde{x}_2)\) does not depend on \(t\). Mathematically, it can be written as:
Additional properties of the proportional hazards model
If \({\lambda}(t|x)= {\lambda}_0(t)\theta(x)\), then:
Theorem 10 (Cumulative hazards are also proportional to \({\Lambda}_0(t)\))\[
\begin{aligned}
{\Lambda}(t|x)
&\stackrel{\text{def}}{=}\int_{u=0}^t {\lambda}(u)du\\
&= \int_{u=0}^t {\lambda}_0(u)\theta(x)du\\
&= \theta(x)\int_{u=0}^t {\lambda}_0(u)du\\
&= \theta(x){\Lambda}_0(t)
\end{aligned}
\]
where \({\Lambda}_0(t) \stackrel{\text{def}}{=}{\Lambda}(t|0) = \int_{u=0}^t {\lambda}_0(u)du\).
Theorem 11 (The logarithms of cumulative hazard should be parallel)\[
\text{log}{\left\{{\Lambda}(t|\tilde{x})\right\}} =\text{log}{\left\{{\Lambda}_0(t)\right\}} + \tilde{x}\cdot \tilde{\beta}
\]
Corollary 4 (linear model for log-negative-log survival)\[
\text{log}{\left\{-\text{log}{\left\{\text{S}(t|\tilde{x})\right\}}\right\}} =
\text{log}{\left\{-\text{log}{\left\{\text{S}_0(t)\right\}}\right\}} + \tilde{x}\cdot \tilde{\beta}
\]
Theorem 12 (Survival functions are exponential multiples of \(\text{S}_0(t)\))\[\text{S}(t|x) = {\left[\text{S}_0(t)\right]}^{\theta(x)}\]
5 Example: Proportional hazards model for the bmt data
Fit the model
[R code]
library(survival)bmt.cox <-coxph(Surv(t2, d3) ~ group, data = bmt)summary(bmt.cox)#> Call:#> coxph(formula = Surv(t2, d3) ~ group, data = bmt)#> #> n= 137, number of events= 83 #> #> coef exp(coef) se(coef) z Pr(>|z|) #> groupLow Risk AML -0.574 0.563 0.287 -2.00 0.046 *#> groupHigh Risk AML 0.383 1.467 0.267 1.43 0.152 #> ---#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1#> #> exp(coef) exp(-coef) lower .95 upper .95#> groupLow Risk AML 0.563 1.776 0.321 0.989#> groupHigh Risk AML 1.467 0.682 0.869 2.478#> #> Concordance= 0.625 (se = 0.03 )#> Likelihood ratio test= 13.4 on 2 df, p=0.001#> Wald test = 13 on 2 df, p=0.001#> Score (logrank) test = 13.8 on 2 df, p=0.001
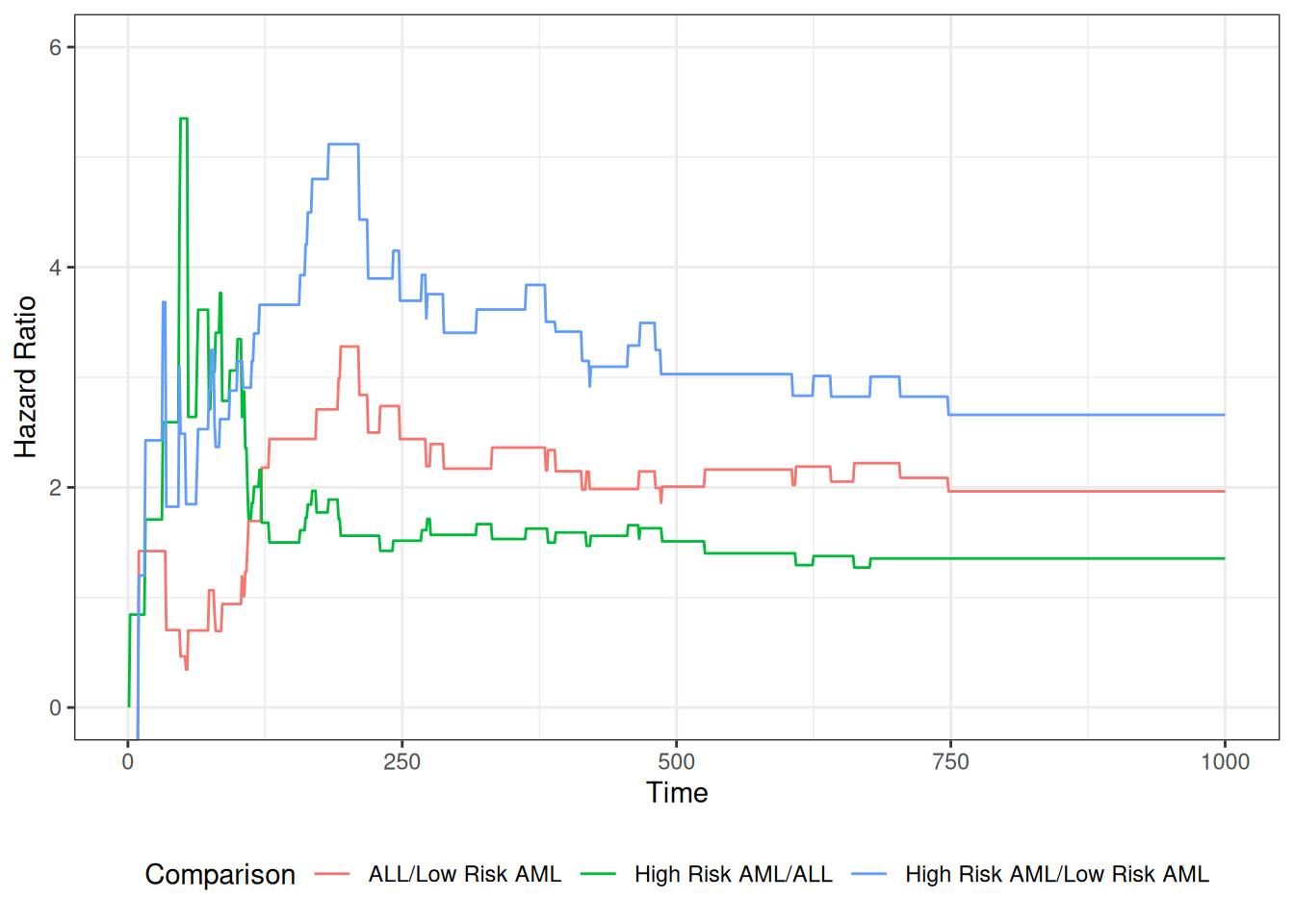
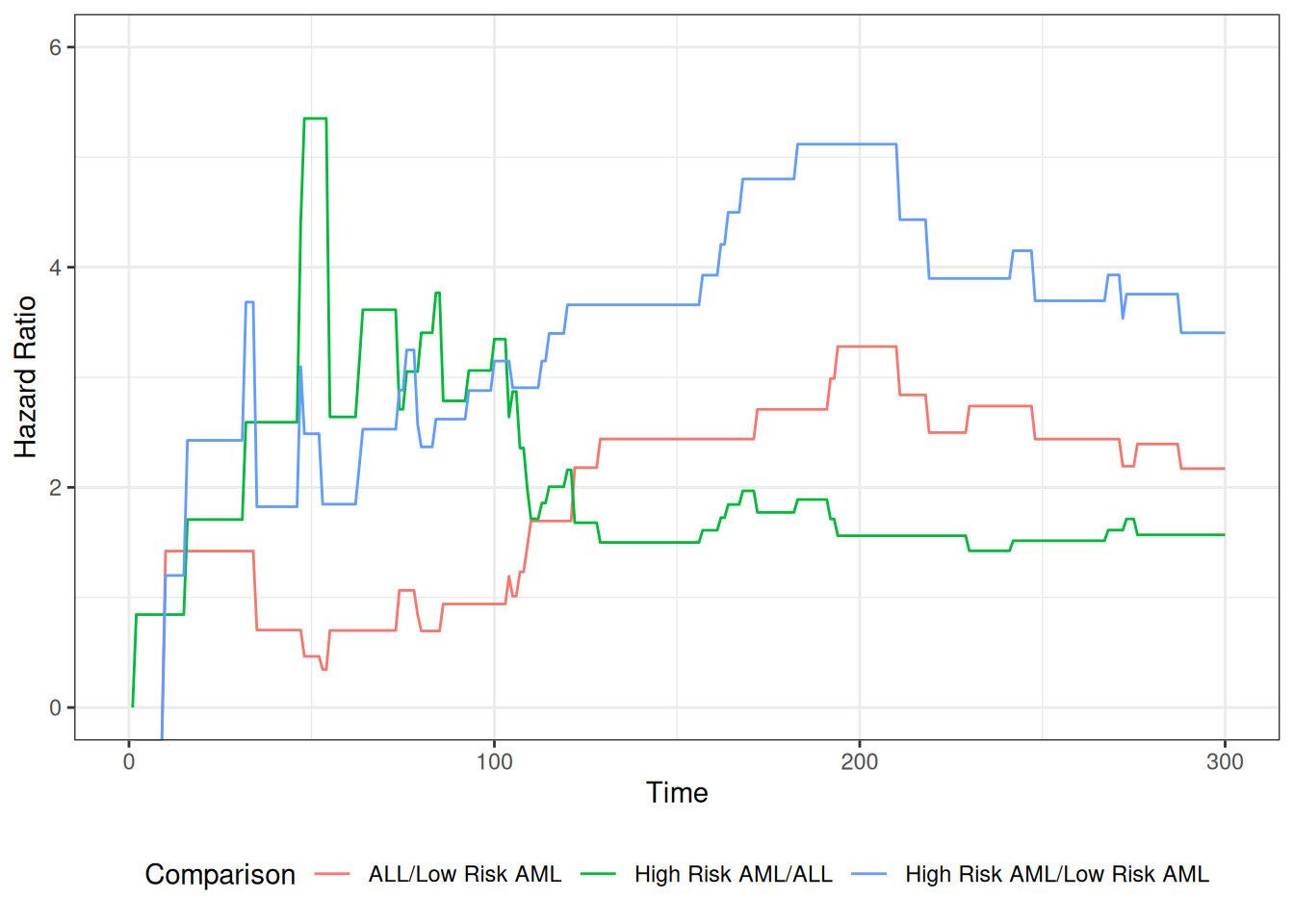
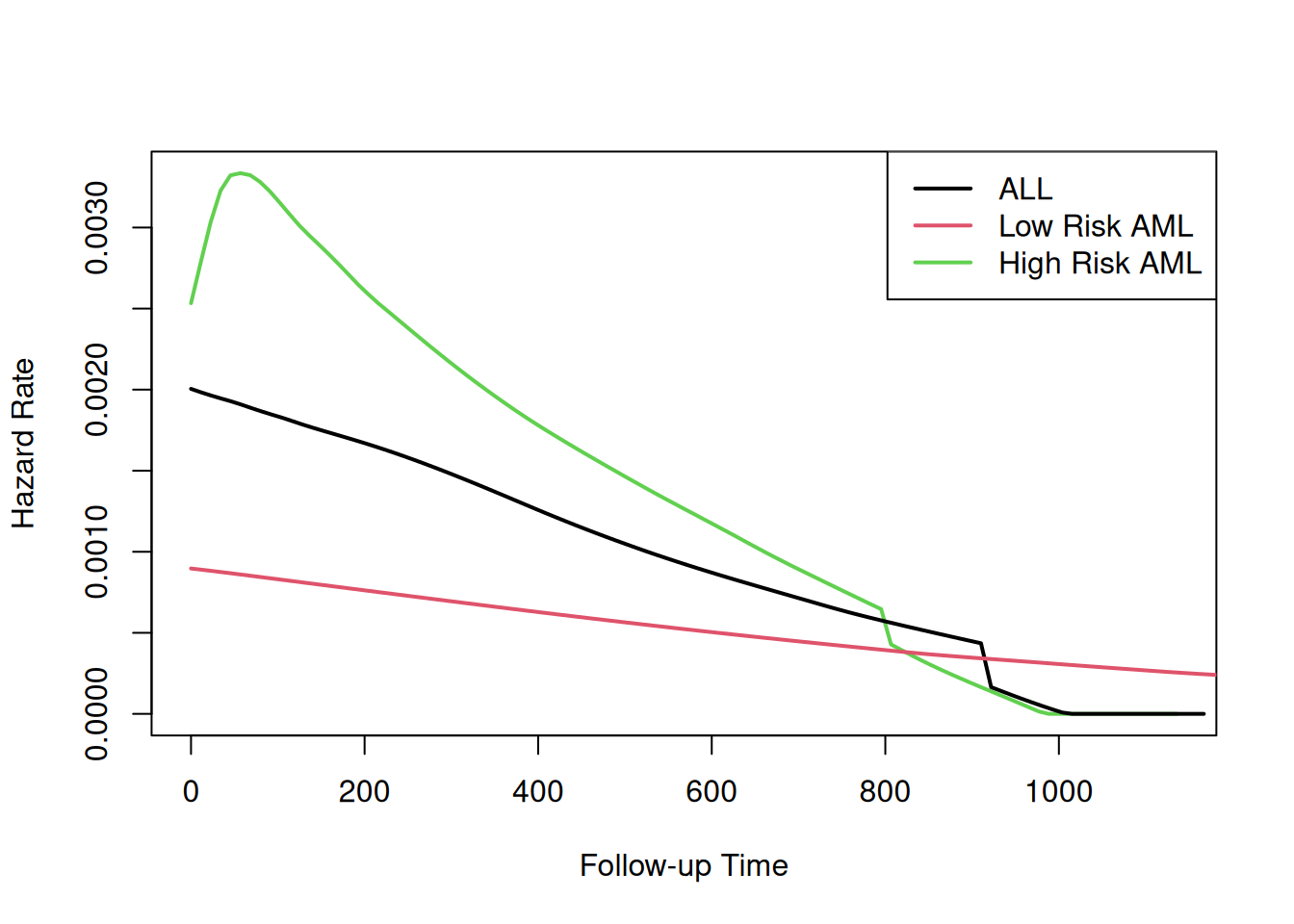
The table provides hypothesis tests comparing groups 2 and 3 to group 1. Group 3 has the highest hazard, so the most significant comparison is not directly shown.
The coefficient 0.3834 is on the log-hazard-ratio scale, as in log-risk-ratio. The next column gives the hazard ratio 1.4673, and a hypothesis (Wald) test.
The (not shown) group 3 vs. group 2 log hazard ratio is 0.3834 + 0.5742 = 0.9576. The hazard ratio is then exp(0.9576) or 2.605.
Inference on all coefficients and combinations can be constructed using coef(bmt.cox) and vcov(bmt.cox) as with logistic and poisson regression.
Concordance is agreement of first failure between pairs of subjects and higher predicted risk between those subjects, omitting non-informative pairs.
The Rsquare value is Cox and Snell’s pseudo R-squared and is not very useful.
Tests for nested models
summary() prints three tests for whether the model with the group covariate is better than the one without
Likelihood ratio test (chi-squared)
Wald test (also chi-squared), obtained by adding the squares of the z-scores
Score = log-rank test, as with comparison of survival functions.
The likelihood ratio test is probably best in smaller samples, followed by the Wald test.
Survival Curves from the Cox Model
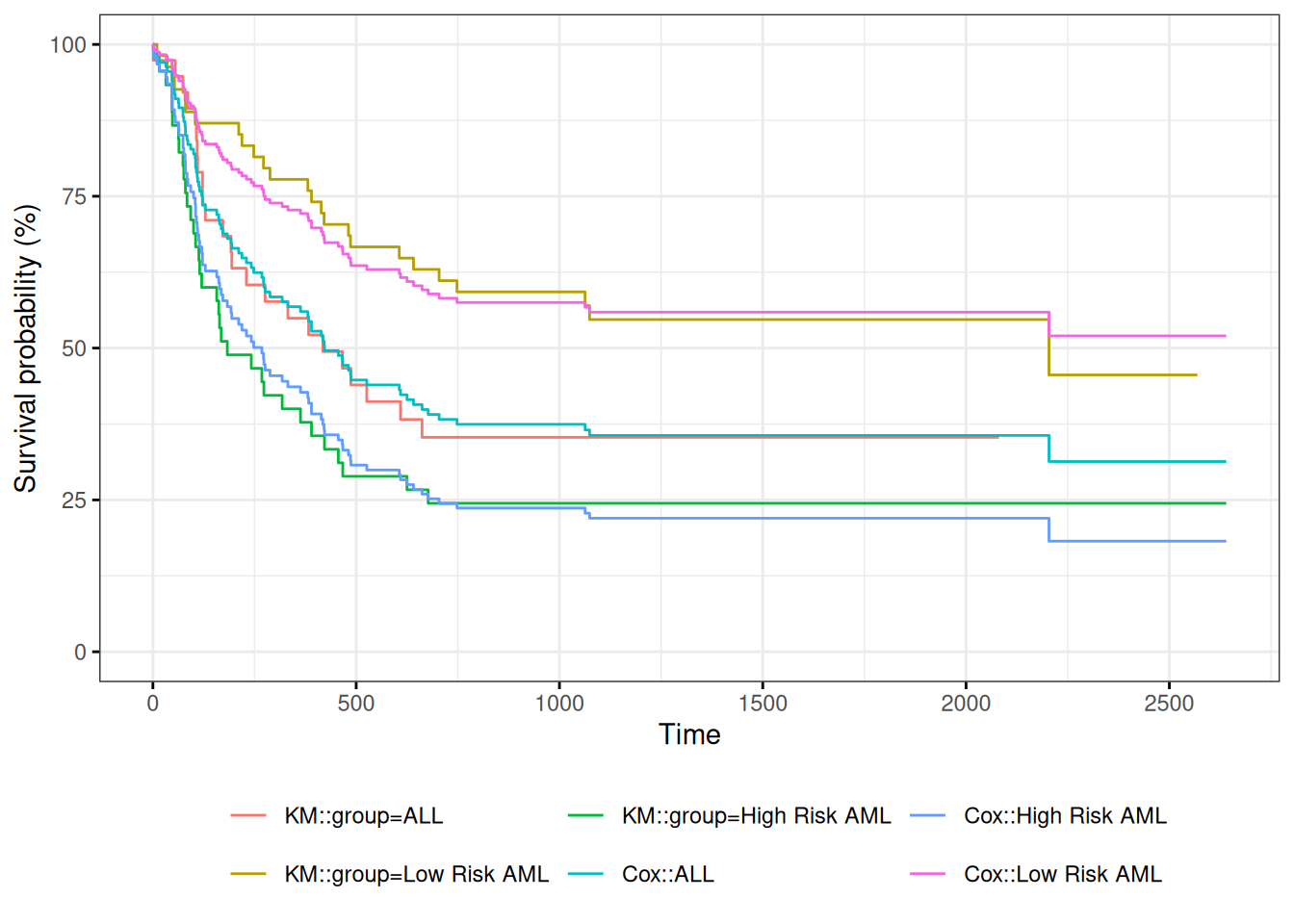
We can take a look at the resulting group-specific curves:
Figure 6: Survival Functions for Three Groups by KM and Cox Model
When we use survfit() with a Cox model, we have to specify the covariate levels we are interested in; the argument newdata should include a data.frame with the same named columns as the predictors in the Cox model and one or more levels of each.
From ?survfit.coxph:
If the newdata argument is missing, a curve is produced for a single “pseudo” subject with covariate values equal to the means component of the fit. The resulting curve(s) almost never make sense, but the default remains due to an unwarranted attachment to the option shown by some users and by other packages. Two particularly egregious examples are factor variables and interactions. Suppose one were studying interspecies transmission of a virus, and the data set has a factor variable with levels (“pig”, “chicken”) and about equal numbers of observations for each. The “mean” covariate level will be 0.5 – is this a flying pig?
Examining survfit
[R code]
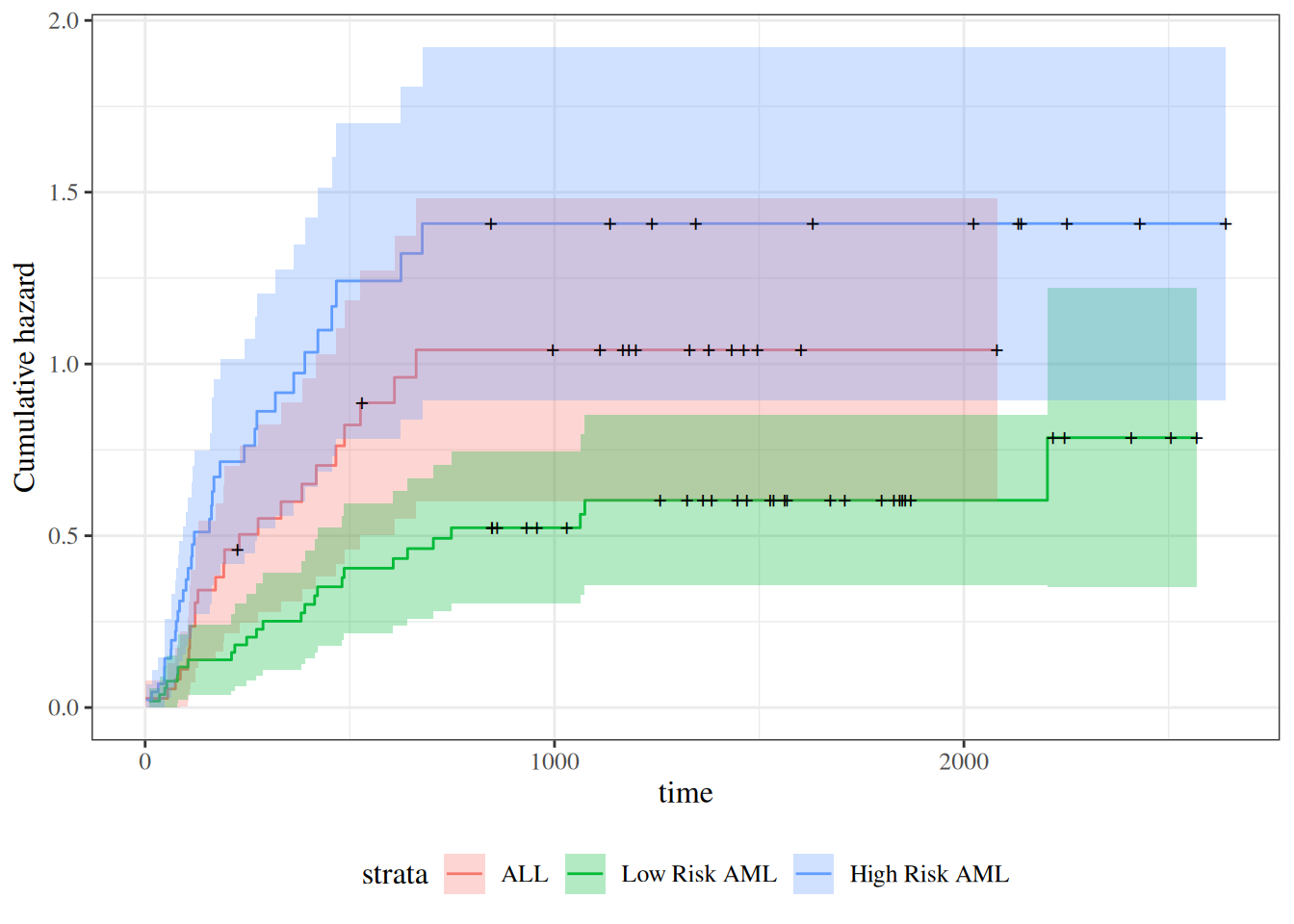
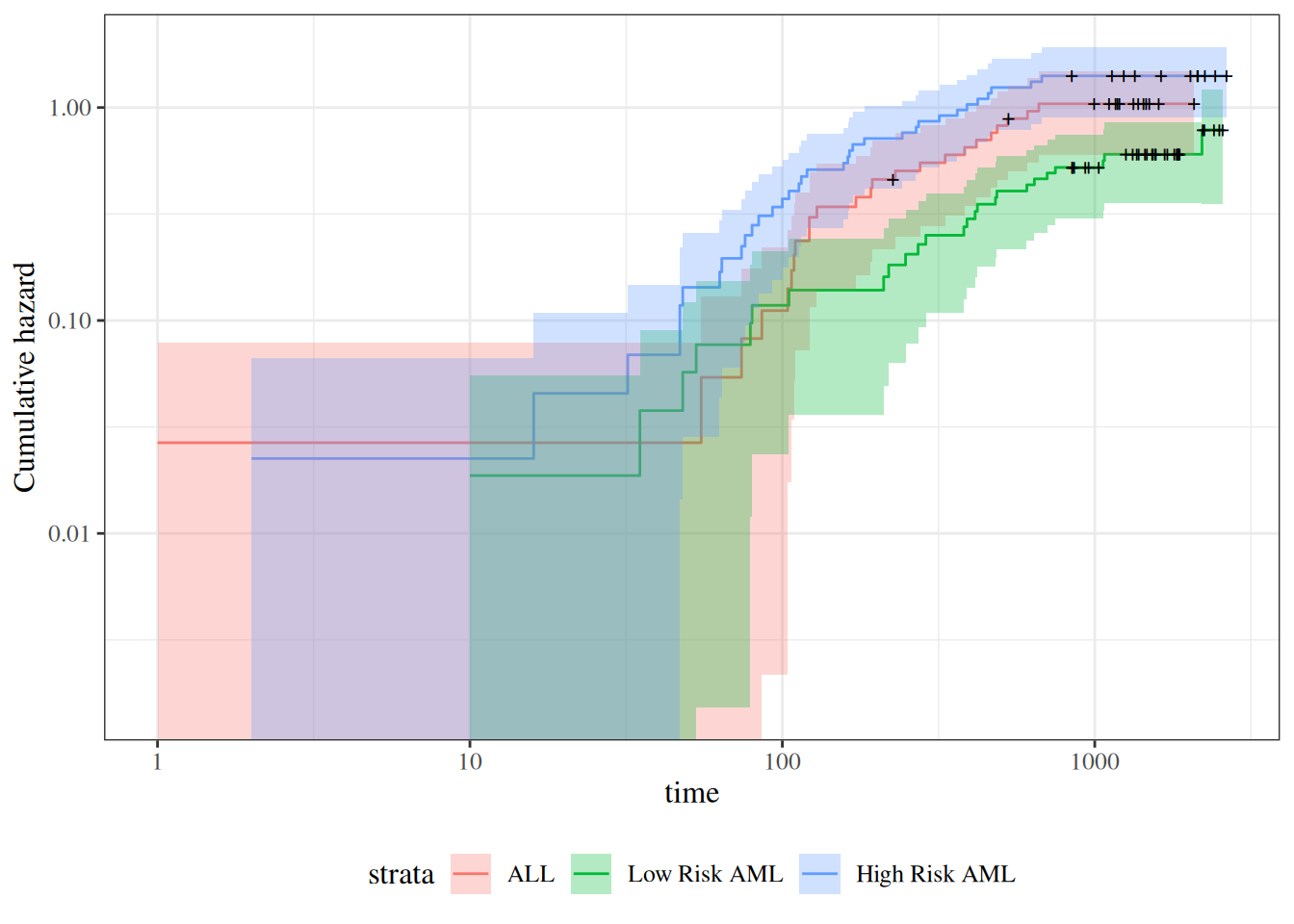
survfit(Surv(t2, d3) ~ group, data = bmt)#> Call: survfit(formula = Surv(t2, d3) ~ group, data = bmt)#> #> n events median 0.95LCL 0.95UCL#> group=ALL 38 24 418 194 NA#> group=Low Risk AML 54 25 2204 704 NA#> group=High Risk AML 45 34 183 115 456
At each time \(t_i\) at which more than one of the subjects has an event, let \(d_i\) be the number of events at that time, \(D_i\) the set of subjects with events at that time, and let \(s_i\) be a covariate vector for an artificial subject obtained by adding up the covariate values for the subjects with an event at time \(t_i\). Let \[\bar\eta_i = \beta_1s_{i1}+\cdots+\beta_ps_{ip}\] and \(\bar\theta_i = \text{exp}{\left\{\bar\eta_i\right\}}\).
Let \(s_i\) be a covariate vector for an artificial subject obtained by adding up the covariate values for the subjects with an event at time \(t_i\). Note that
This method is equivalent to treating each event as distinct and using the non-ties formula. It works best when the number of ties is small. It is the default in many statistical packages, including PROC PHREG in SAS.
Efron’s method for ties
The other common method is Efron’s, which is the default in R.
\[L(\beta|T)=
\prod_i \frac{\bar\theta_i}{\prod_{j=1}^{d_i}[\sum_{k \in R(t_i)} \theta_k-\frac{j-1}{d_i}\sum_{k \in D_i} \theta_k]}\] This is closer to the exact discrete partial likelihood when there are many ties.
The third option in R (and an option also in SAS as discrete) is the “exact” method, which is the same one used for matched logistic regression.
Example: Breslow’s method
Suppose as an example we have a time \(t\) where there are 20 individuals at risk and three failures. Let the three individuals have risk parameters \(\theta_1, \theta_2, \theta_3\) and let the sum of the risk parameters of the remaining 17 individuals be \(\theta_R\). Then the factor in the partial likelihood at time \(t\) using Breslow’s method is
as the risk set got smaller with each failure. The exact method roughly averages the results for the six possible orderings of the failures.
Example: Efron’s method
But we don’t know the order they failed in, so instead of reducing the denominator by one risk coefficient each time, we reduce it by the same fraction. This is Efron’s method.
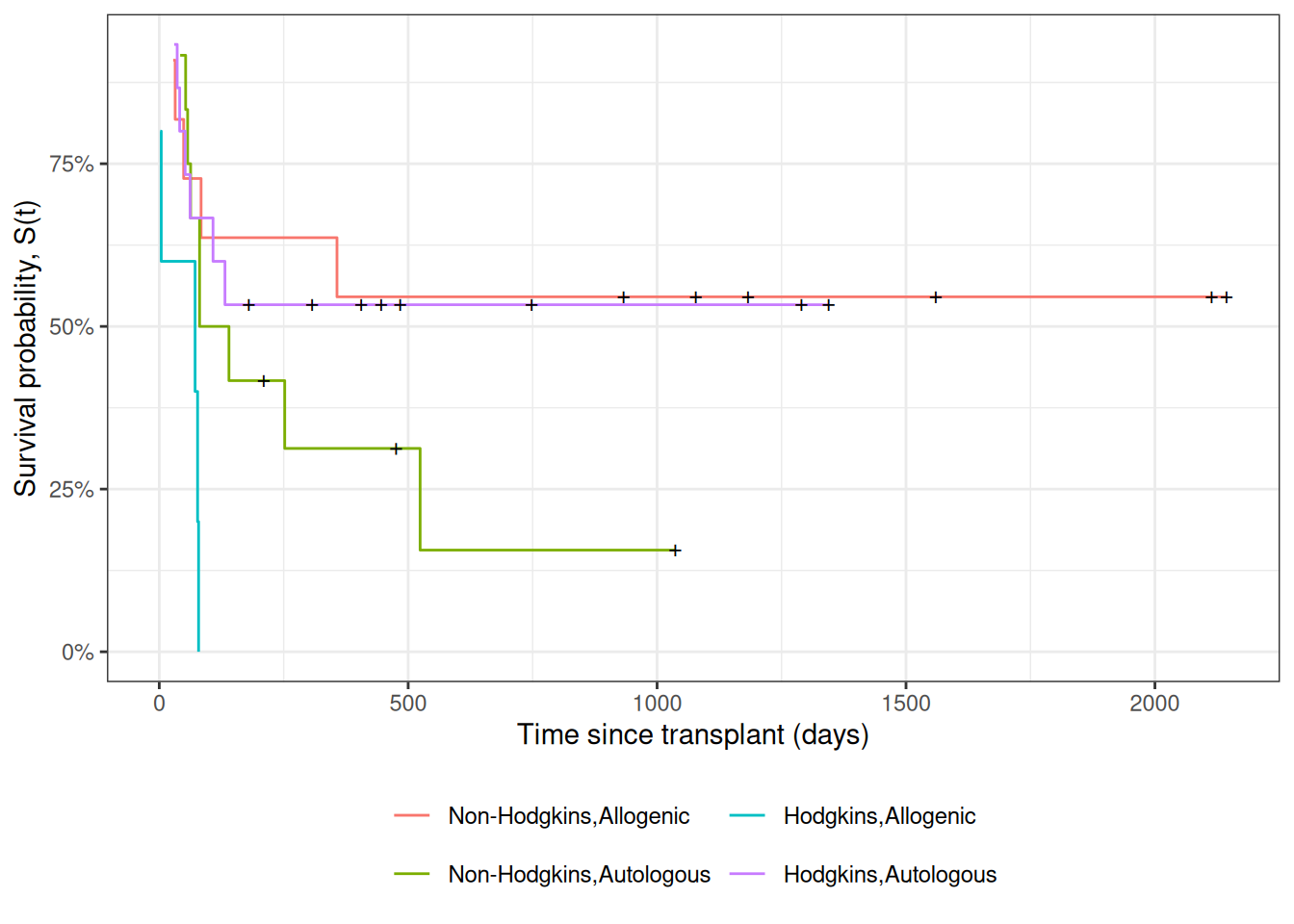
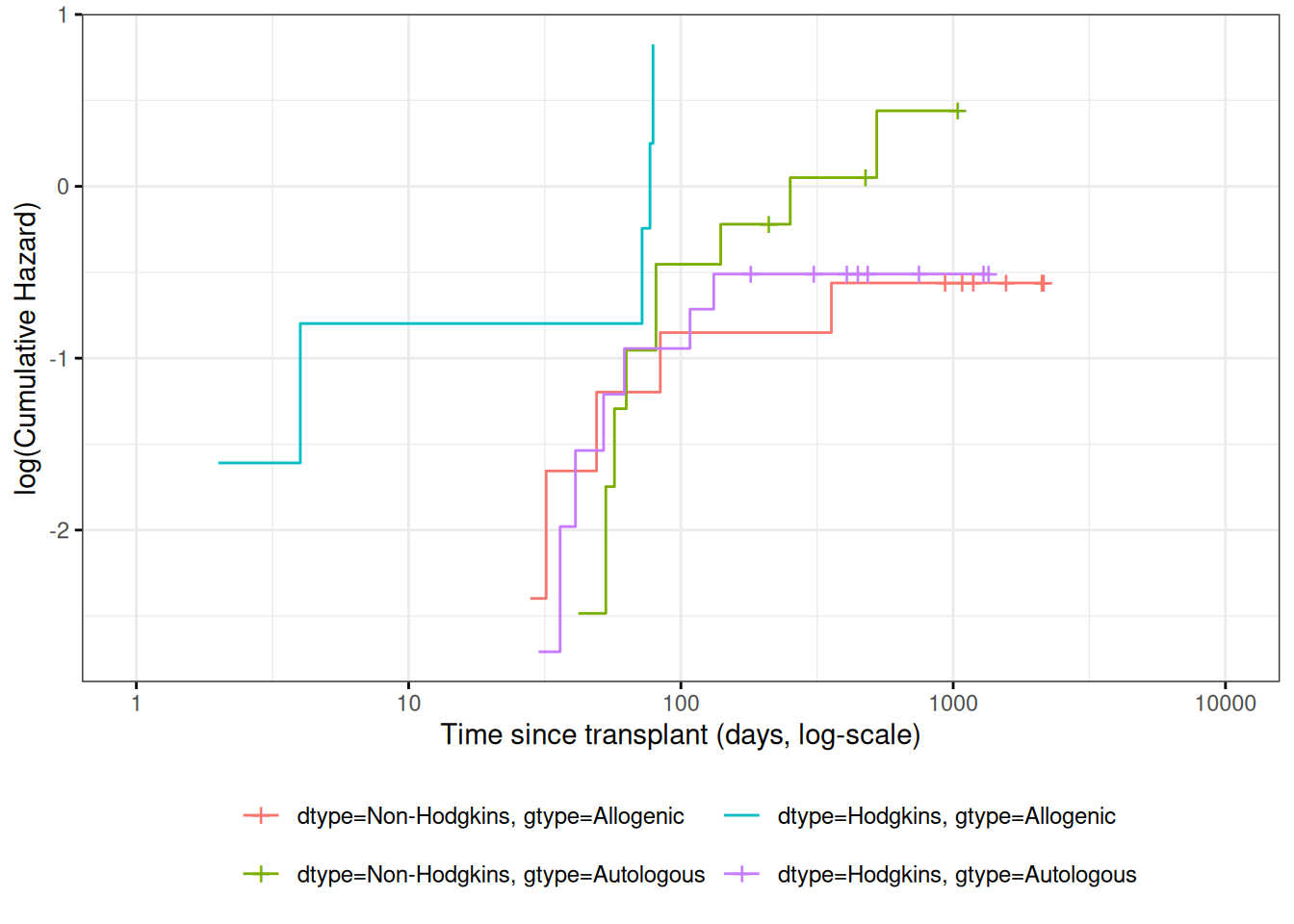
Figure 7: Kaplan-Meier Survival Curves for HOD/NHL and Allo/Auto Grafts
Observed and expected survival curves
[R code]
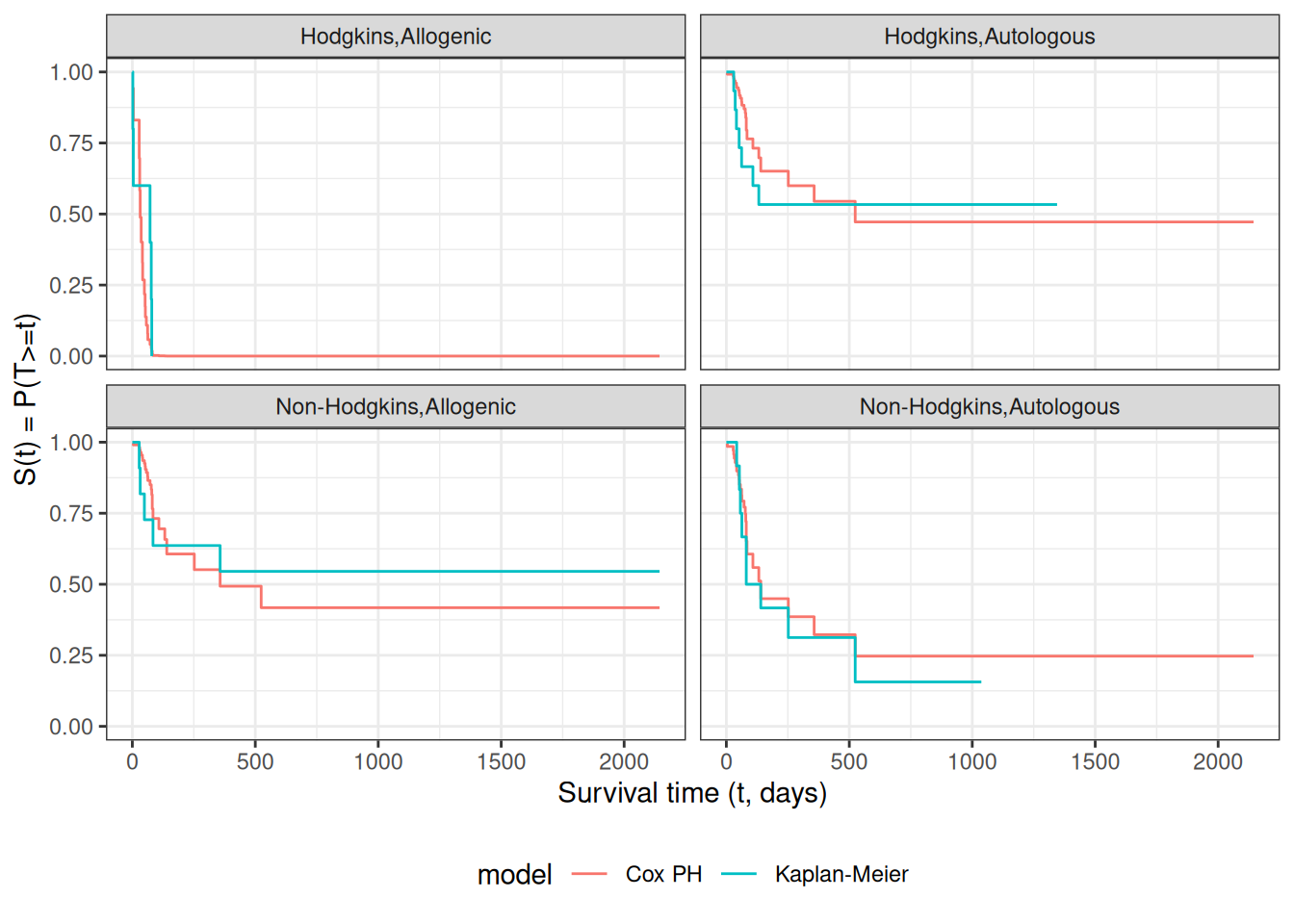
# we need to create a tibble of covariate patterns;# we will set score and wtime to mean values for disease and graft types:means = hodg2 |>summarize(.by =c(dtype, gtype), score =mean(score), wtime =mean(wtime)) |>arrange(dtype, gtype) |>mutate(strata =paste(dtype, gtype, sep =",")) |>as.data.frame() # survfit.coxph() will use the rownames of its `newdata`# argument to label its output:rownames(means) = means$stratacox_model = hodg.cox1 |>survfit(data = hodg2, # ggsurvplot() will need thisnewdata = means)
[R code]
# I couldn't find a good function to reformat `cox_model` for ggplot, # so I made my own:stack_surv_ph =function(cox_model){ cox_model$surv |>as_tibble() |>mutate(time = cox_model$time) |>pivot_longer(cols =-time,names_to ="strata",values_to ="surv") |>mutate(cumhaz =-log(surv),model ="Cox PH")}km_and_cph = km_model |>fortify(surv.connect =TRUE) |>mutate(strata =trimws(strata),model ="Kaplan-Meier",cumhaz =-log(surv)) |>bind_rows(stack_surv_ph(cox_model))
[R code]
km_and_cph |>ggplot(aes(x = time, y = surv, col = model)) +geom_step() +facet_wrap(~strata) +theme_bw() +ylab("S(t) = P(T>=t)") +xlab("Survival time (t, days)") +theme(legend.position ="bottom")
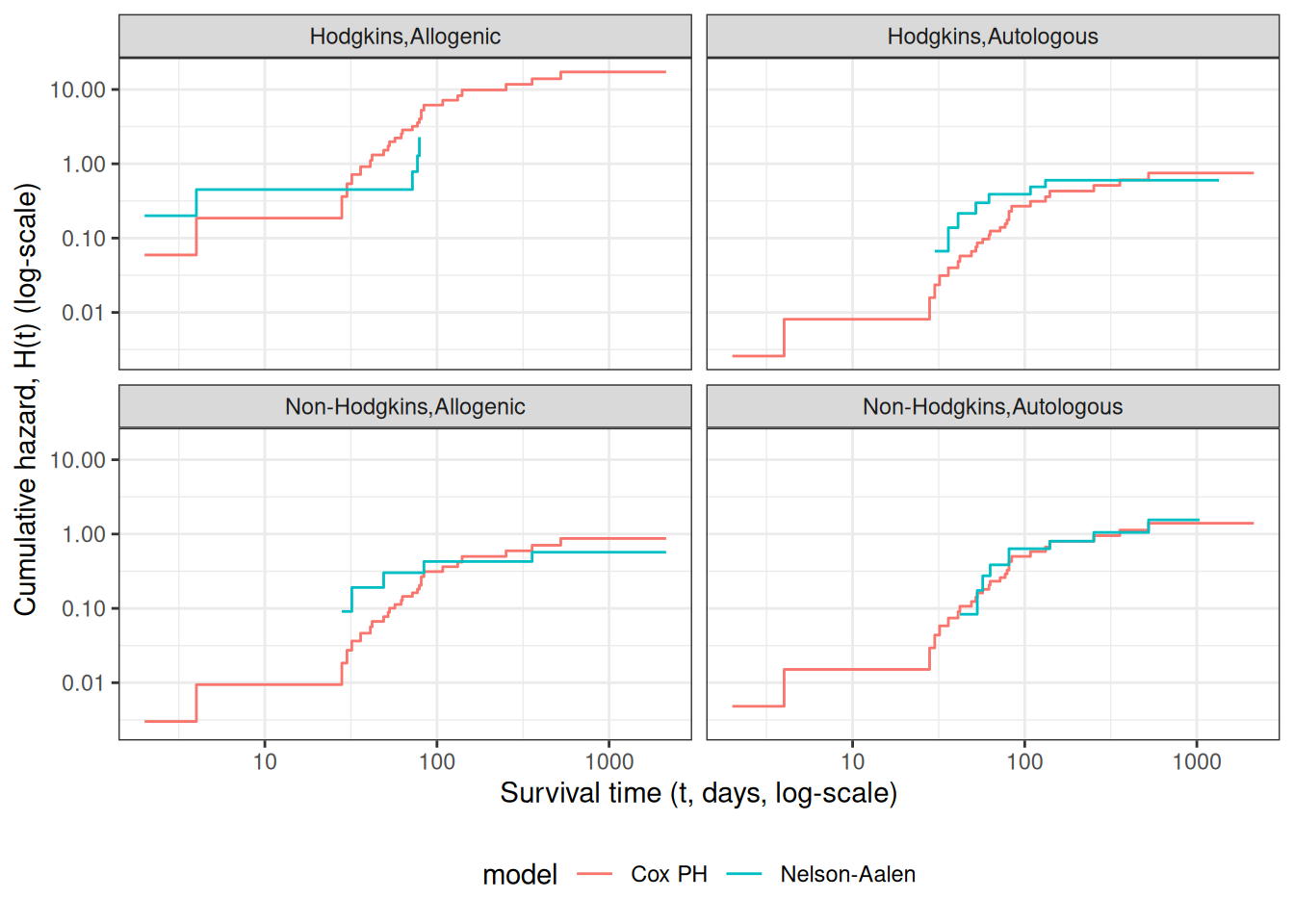
Observed and expected survival curves for bmt data
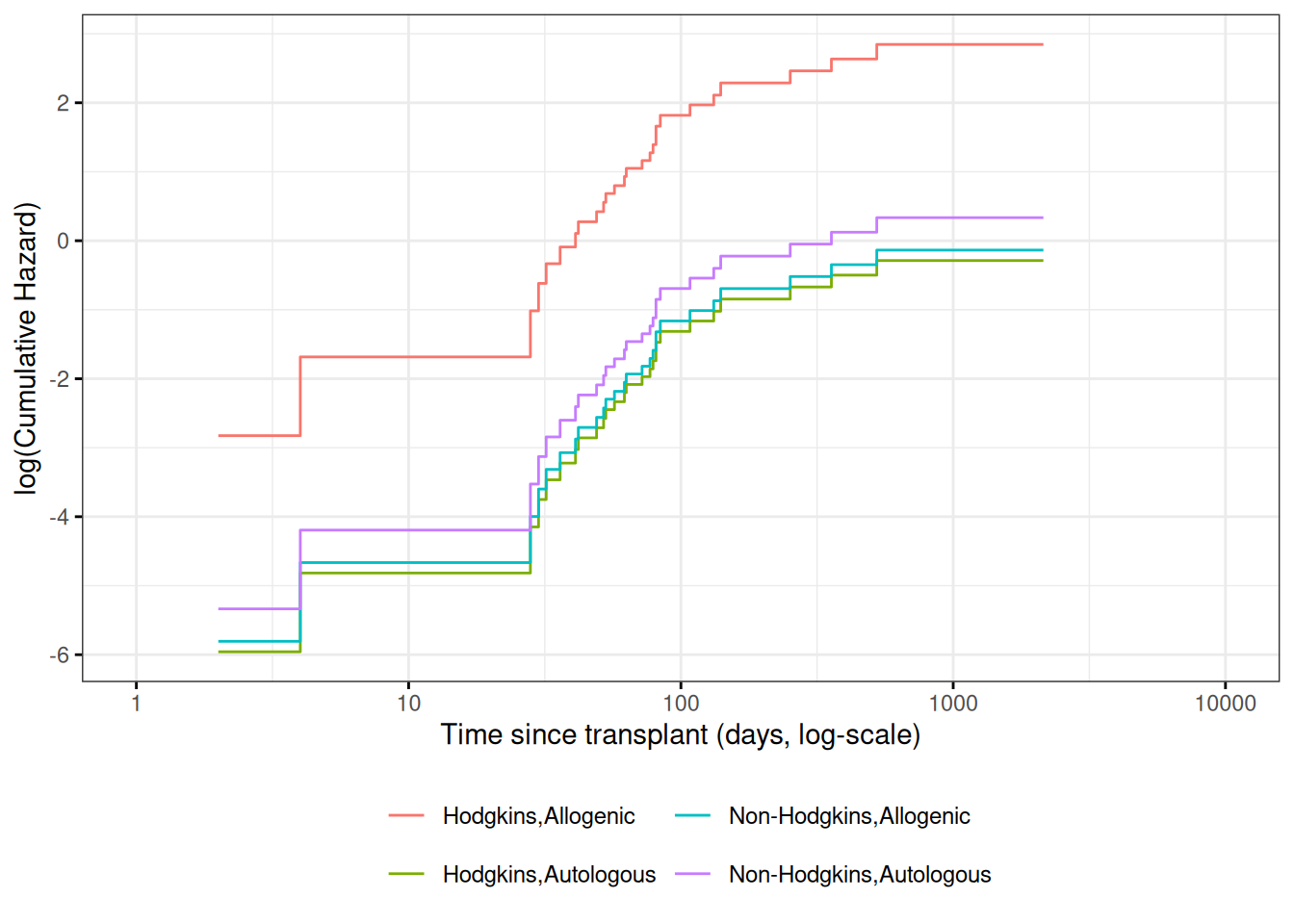
Cumulative hazard (log-scale) curves
Also known as “complementary log-log (clog-log) survival curves”.
\(\hat y_i\) estimates the conditional mean of \(y_i\) given the covariate values \(\tilde{x}_i\). This together with the prediction error says that we are predicting the distribution of values of \(y\).
Review: Residuals in Linear Regression
The usual residual is \(r_i=y_i-\hat y_i\), the difference between the actual value of \(y\) and a prediction of its mean.
The residuals are also the quantities the sum of whose squares is being minimized by the least squares/MLE estimation.
Predictions and Residuals in survival models
In survival analysis, the equivalent of \(y_i\) is the event time \(t_i\), which is unknown for the censored observations.
The expected event time can be tricky to calculate:
The nature of time-to-event data results in very wide prediction intervals:
Suppose a cancer patient is predicted to have a mean lifetime of 5 years after diagnosis and suppose the distribution is exponential.
If we want a 95% interval for survival, the lower end is at the 0.025 percentage point of the exponential which is qexp(.025, rate = 1/5) = 0.126589 years, or 1/40 of the mean lifetime.
The upper end is at the 0.975 point which is qexp(.975, rate = 1/5) = 18.444397 years, or 3.7 times the mean lifetime.
Saying that the survival time is somewhere between 6 weeks and 18 years does not seem very useful, but it may be the best we can do.
For survival analysis, something is like a residual if it is small when the model is accurate or if the accumulation of them is in some way minimized by the estimation algorithm, but there is no exact equivalence to linear regression residuals.
And if there is, they are mostly quite large!
Types of Residuals in Time-to-Event Models
It is often hard to make a decision from graph appearances, though the process can reveal much.
Some diagnostic tests are based on residuals as with other regression methods:
Schoenfeld residuals (via cox.zph) for proportionality
Cox-Snell residuals for goodness of fit (Section 6.4)
martingale residuals for non-linearity
dfbeta for influence.
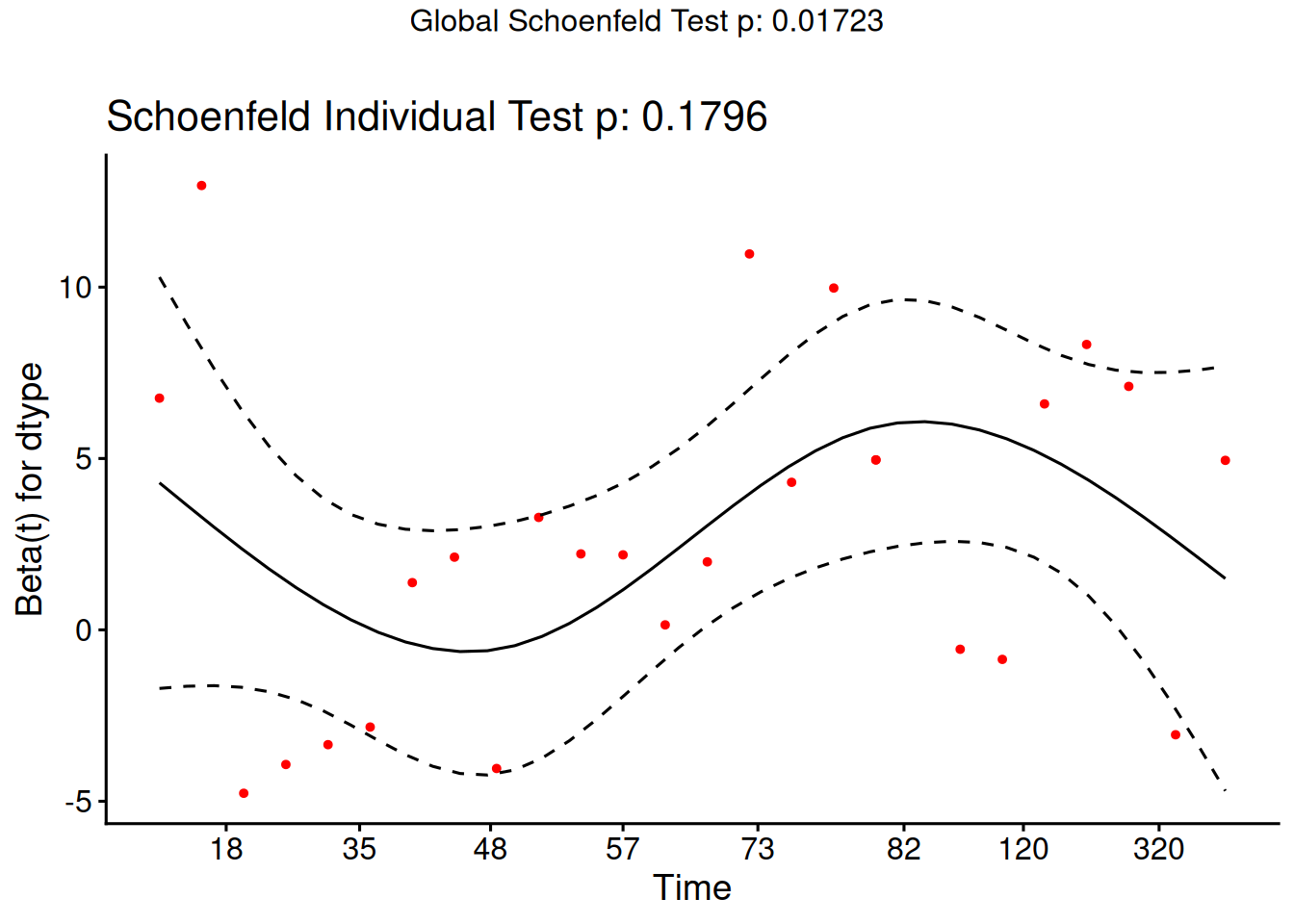
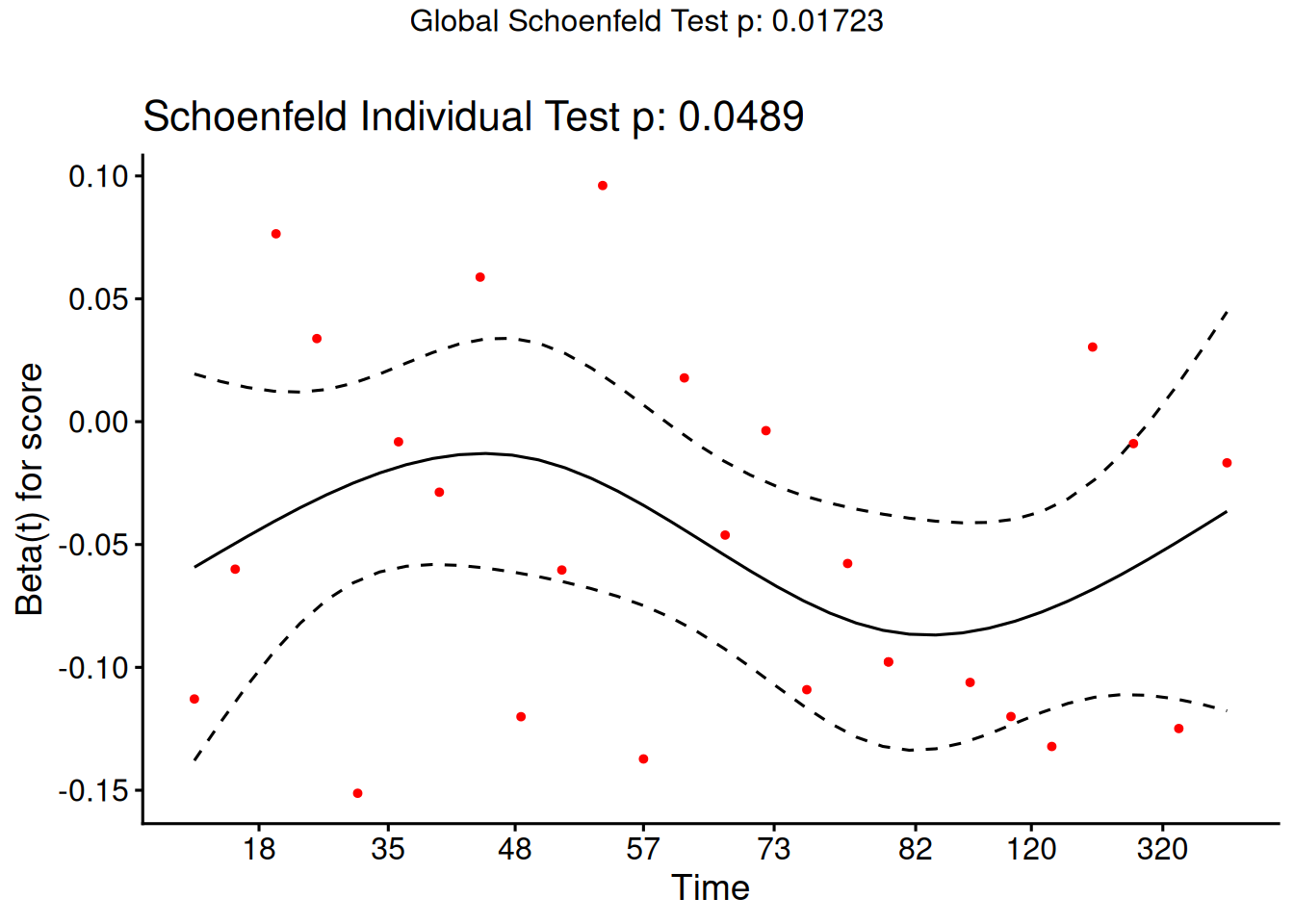
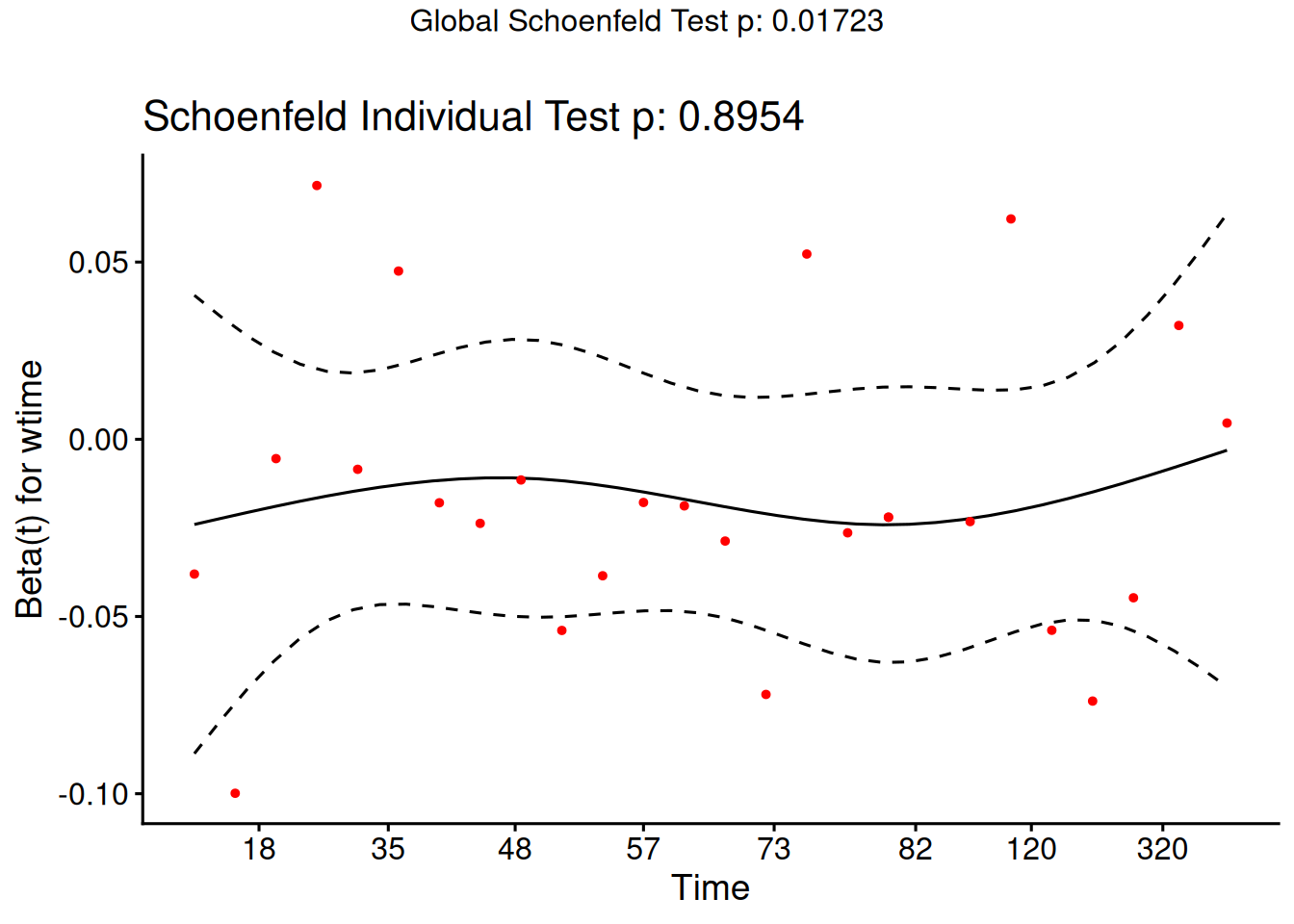
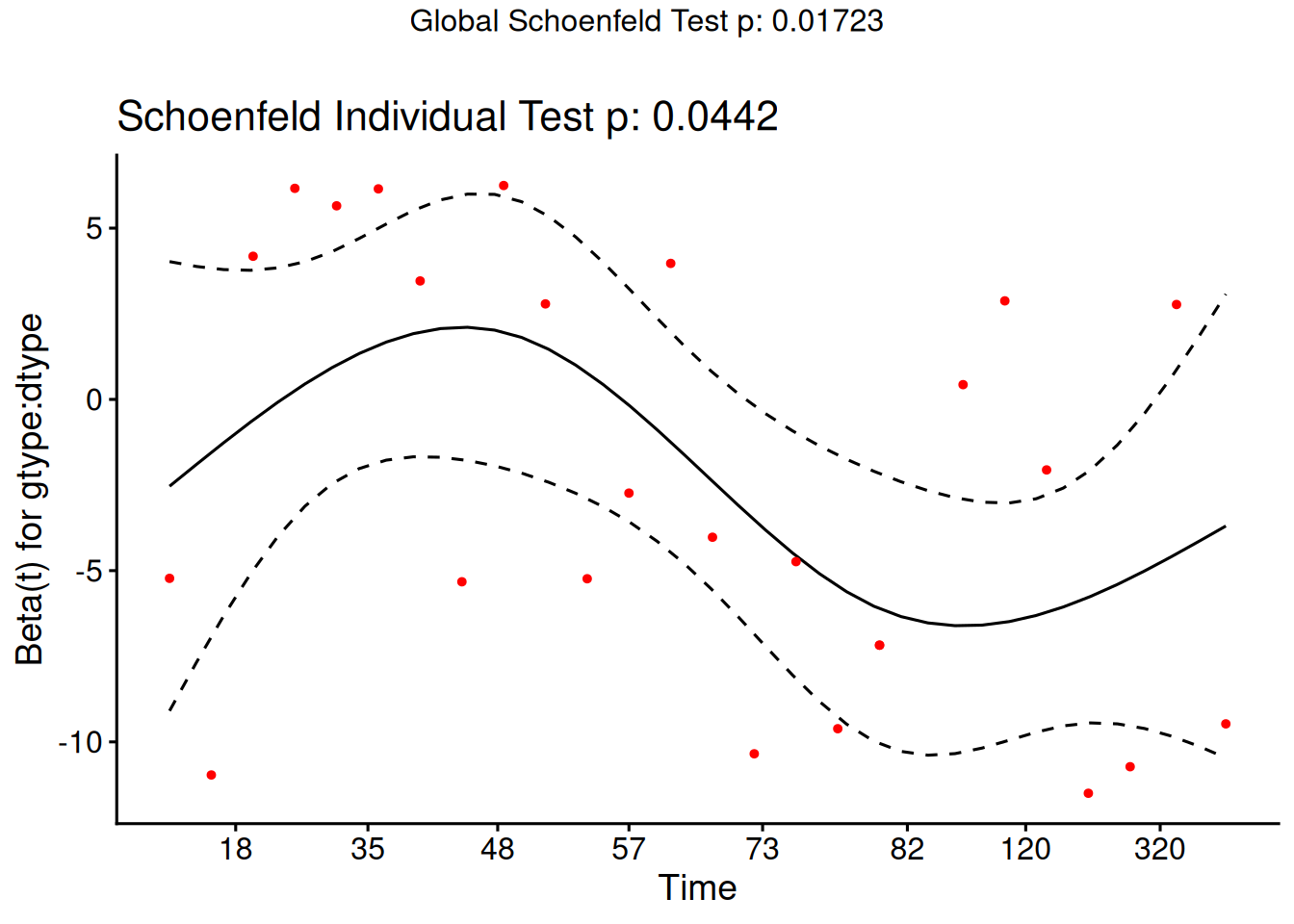
Schoenfeld residuals
There is a Schoenfeld residual for each subject \(i\) with an event (not censored) and for each predictor \(x_{k}\).
At the event time \(t\) for that subject, there is a risk set \(R\), and each subject \(j\) in the risk set has a risk coefficient \(\theta_j\) and also a value \(x_{jk}\) of the predictor.
The Schoenfeld residual is the difference between \(x_{ik}\) and the risk-weighted average of all the \(x_{jk}\) over the risk set.
This residual measures how typical the individual subject is with respect to the covariate at the time of the event. Since subjects should fail more or less uniformly according to risk, the Schoenfeld residuals should be approximately level over time, not increasing or decreasing.
We can test this with the correlation with time on some scale, which could be the time itself, the log time, or the rank in the set of failure times.
The default is to use the KM curve as a transform, which is similar to the rank but deals better with censoring.
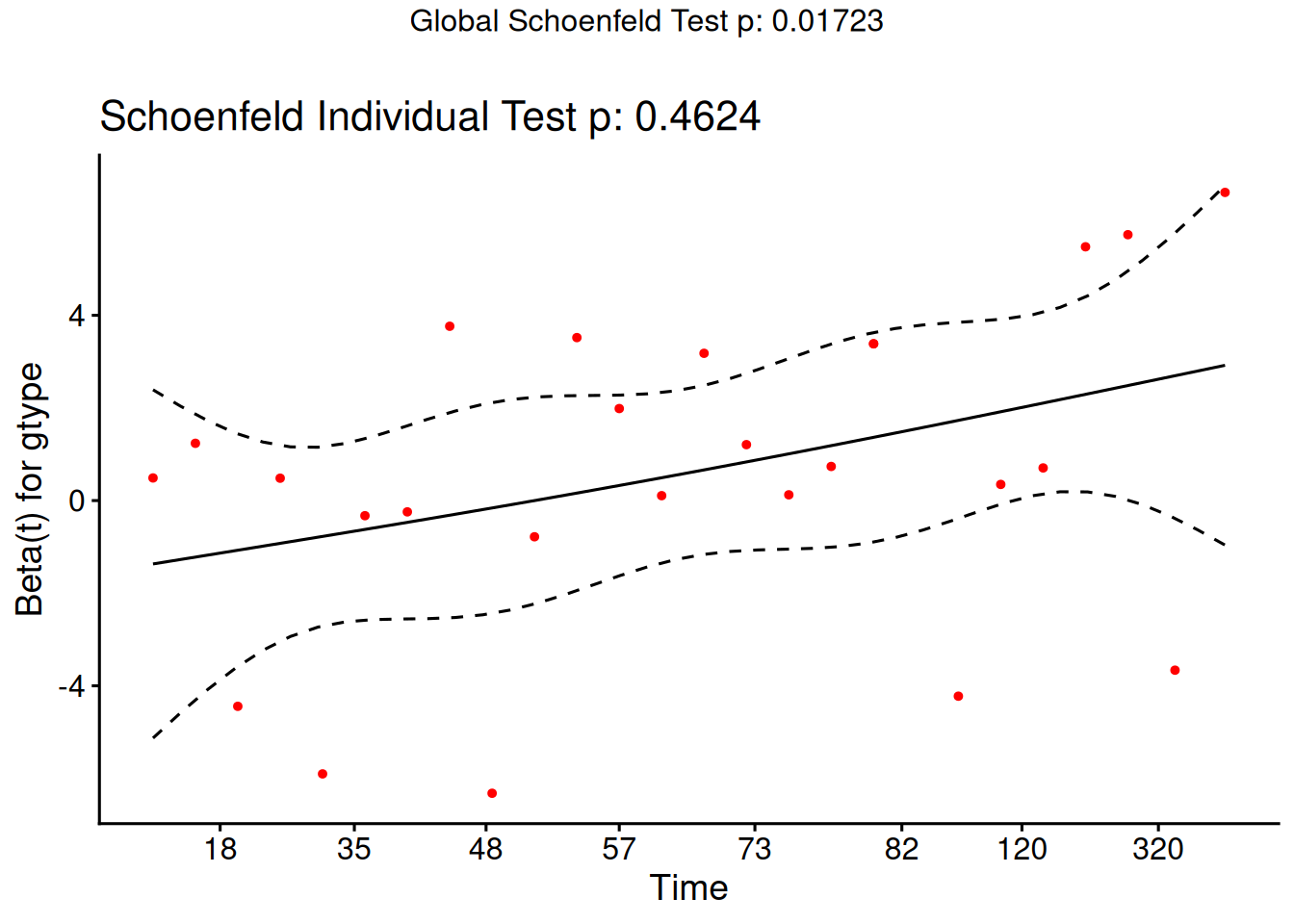
The cox.zph() function implements a score test proposed in Grambsch and Therneau (1994).
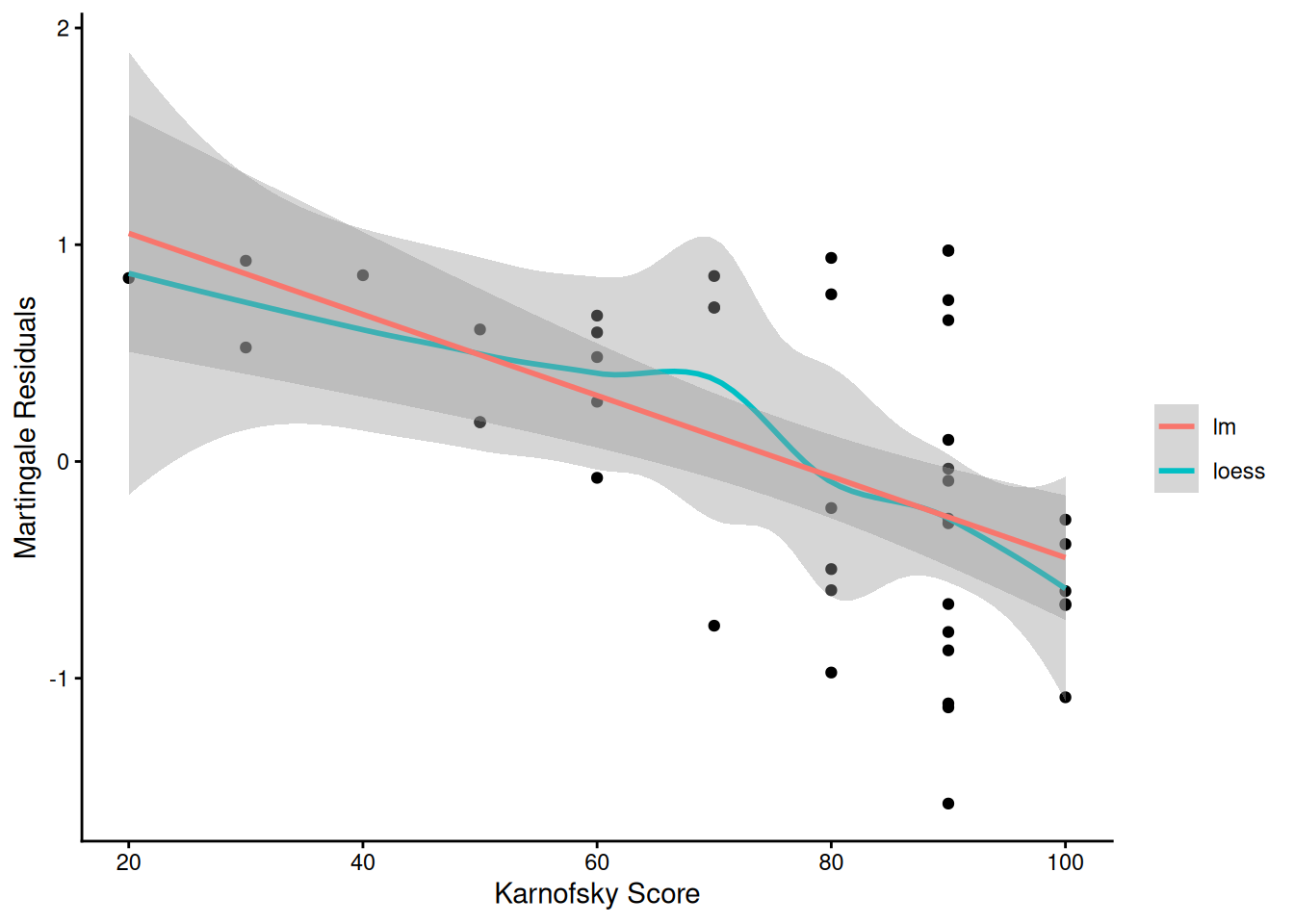
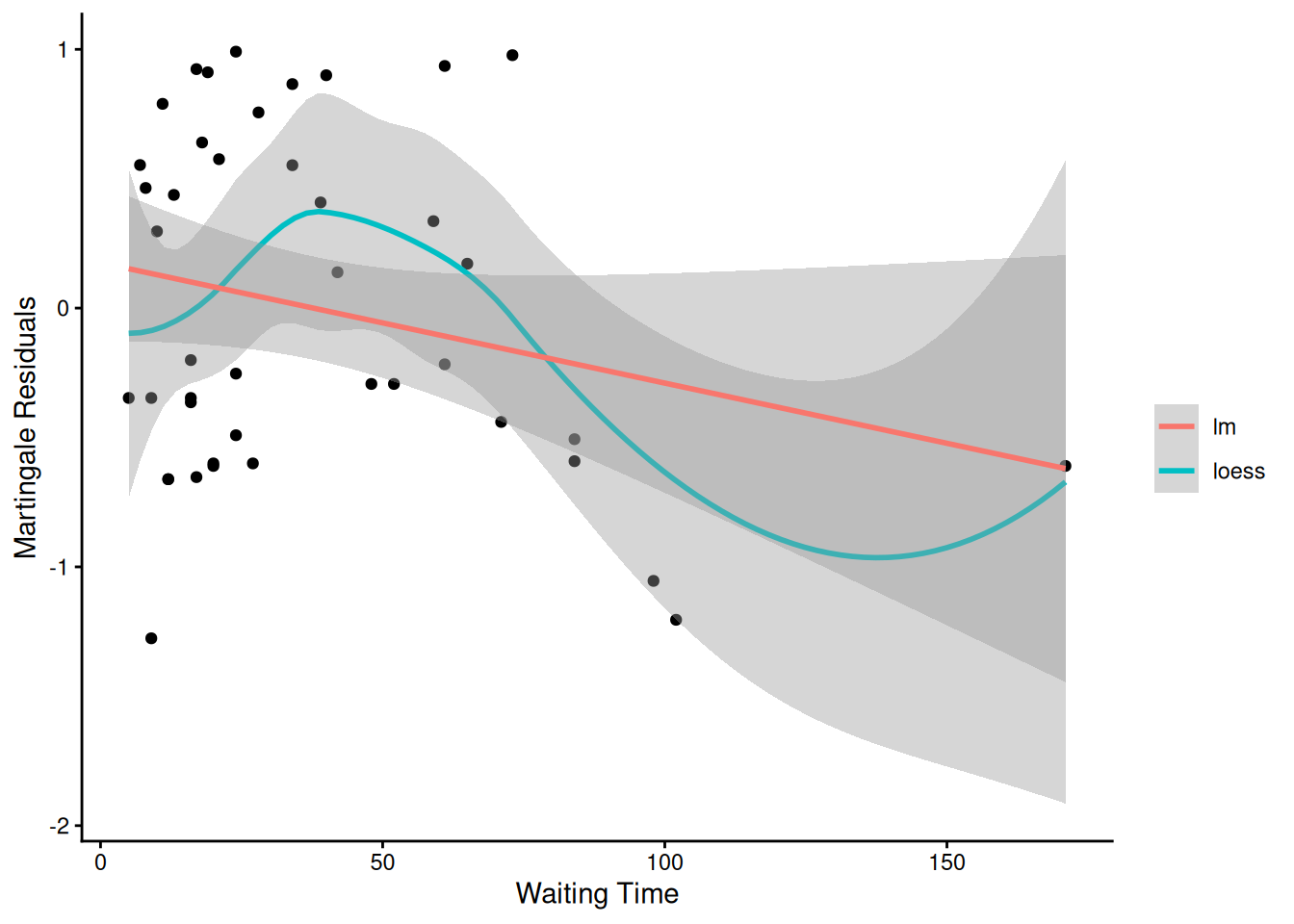
From the correlation test, the Karnofsky score and the interaction with graft type disease type induce modest but statistically significant non-proportionality.
The sample size here is relatively small (26 events in 43 subjects). If the sample size is large, very small amounts of non-proportionality can induce a significant result.
As time goes on, autologous grafts are over-represented at their own event times, but those from HOD patients become less represented.
Both the statistical tests and the plots are useful.
6.4 Goodness of Fit using the Cox-Snell Residuals
(references: Klein and Moeschberger (2003), §11.2, and Dobson and Barnett (2018), §10.6)
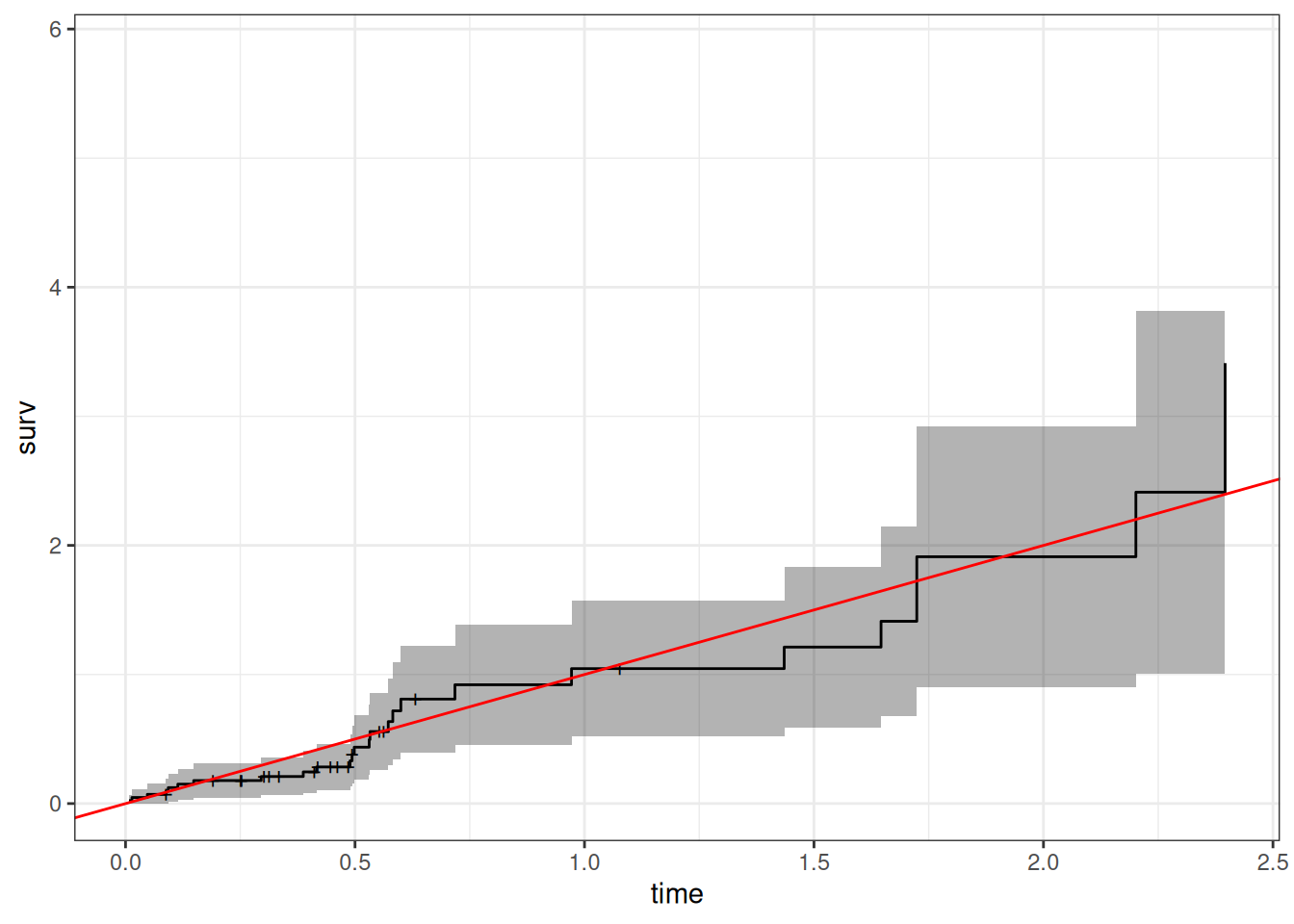
hodg2 = hodg2 |>mutate(cs =predict(hodg.cox1, type ="expected"))surv.csr =survfit(data = hodg2,formula =Surv(time = cs, event = delta =="dead") ~1,type ="fleming-harrington")autoplot(surv.csr, fun ="cumhaz") +geom_abline(aes(intercept =0, slope =1), col ="red") +theme_bw()
Figure 11: Cumulative Hazard of Cox-Snell Residuals
The line with slope 1 and intercept 0 fits the curve relatively well, so we don’t see lack of fit using this procedure.
6.5 Martingale Residuals
The martingale residuals are a slight modification of the Cox-Snell residuals. If the censoring indicator is \(\delta_i\), then \[r^M_i=\delta_i-r^{CS}_i\] These residuals can be interpreted as an estimate of the excess number of events seen in the data but not predicted by the model. We will use these to examine the functional forms of continuous covariates.
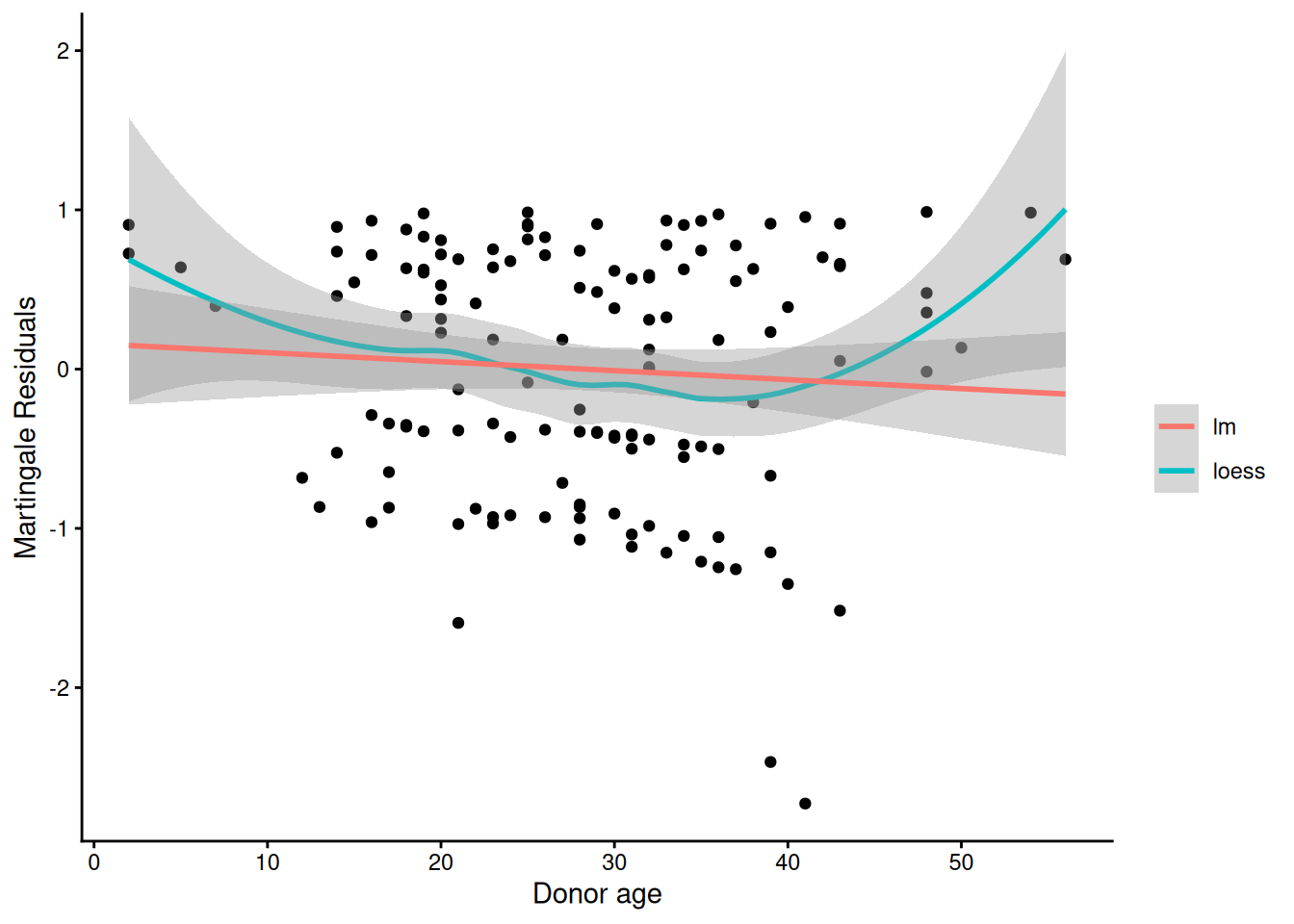
Using Martingale Residuals
Martingale residuals can be used to examine the functional form of a numeric variable.
We fit the model without that variable and compute the martingale residuals.
We then plot these martingale residuals against the values of the variable.
We can see curvature, or a possible suggestion that the variable can be discretized.
Let’s use this to examine the score and wtime variables in the wtime data set.
Model summary table with waiting time on continuous scale
[R code]
hodg.cox2 |>drop1(test="Chisq")
Model summary table with dichotomized waiting time
The new model has better (lower) AIC.
6.6 Checking for Outliers and Influential Observations
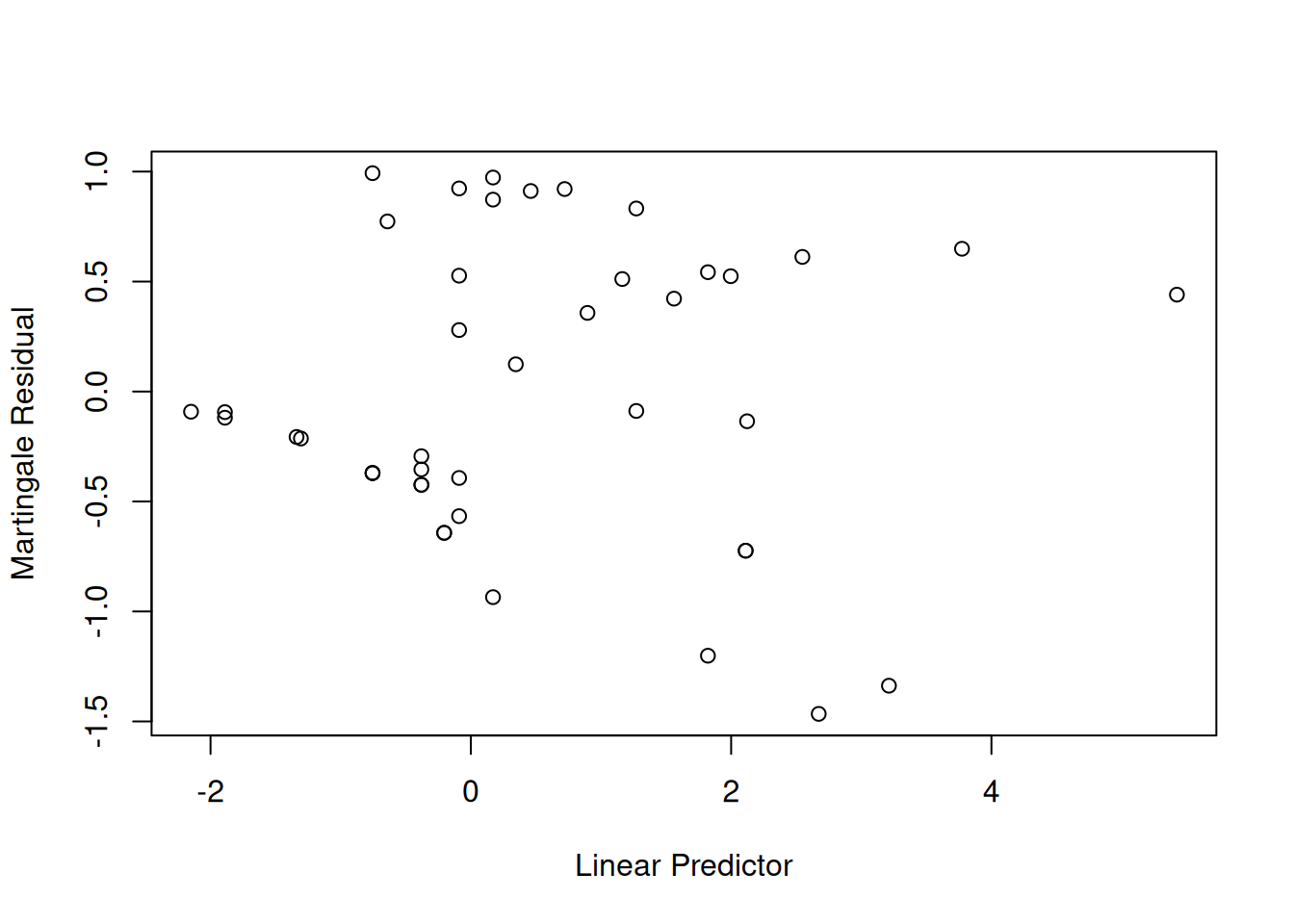
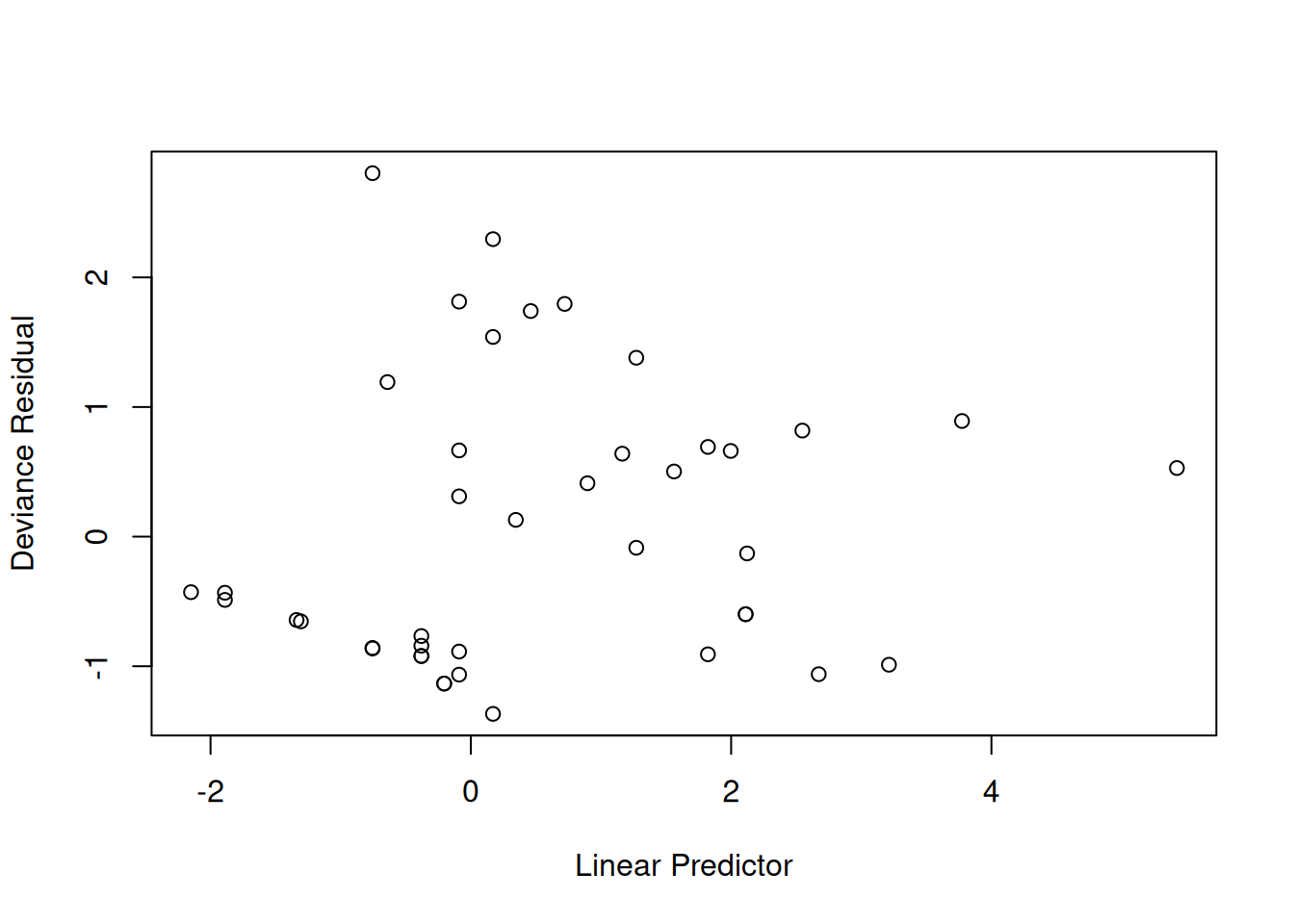
We will check for outliers using the deviance residuals. The martingale residuals show excess events or the opposite, but highly skewed, with the maximum possible value being 1, but the smallest value can be very large negative. Martingale residuals can detect unexpectedly long-lived patients, but patients who die unexpectedly early show up only in the deviance residual. Influence will be examined using dfbeta in a similar way to linear regression, logistic regression, or Poisson regression.
The two largest deviance residuals are observations 1 and 29. Worth examining.
dfbeta
dfbeta is the approximate change in the coefficient vector if that observation were dropped
dfbetas is the approximate change in the coefficients, scaled by the standard error for the coefficients.
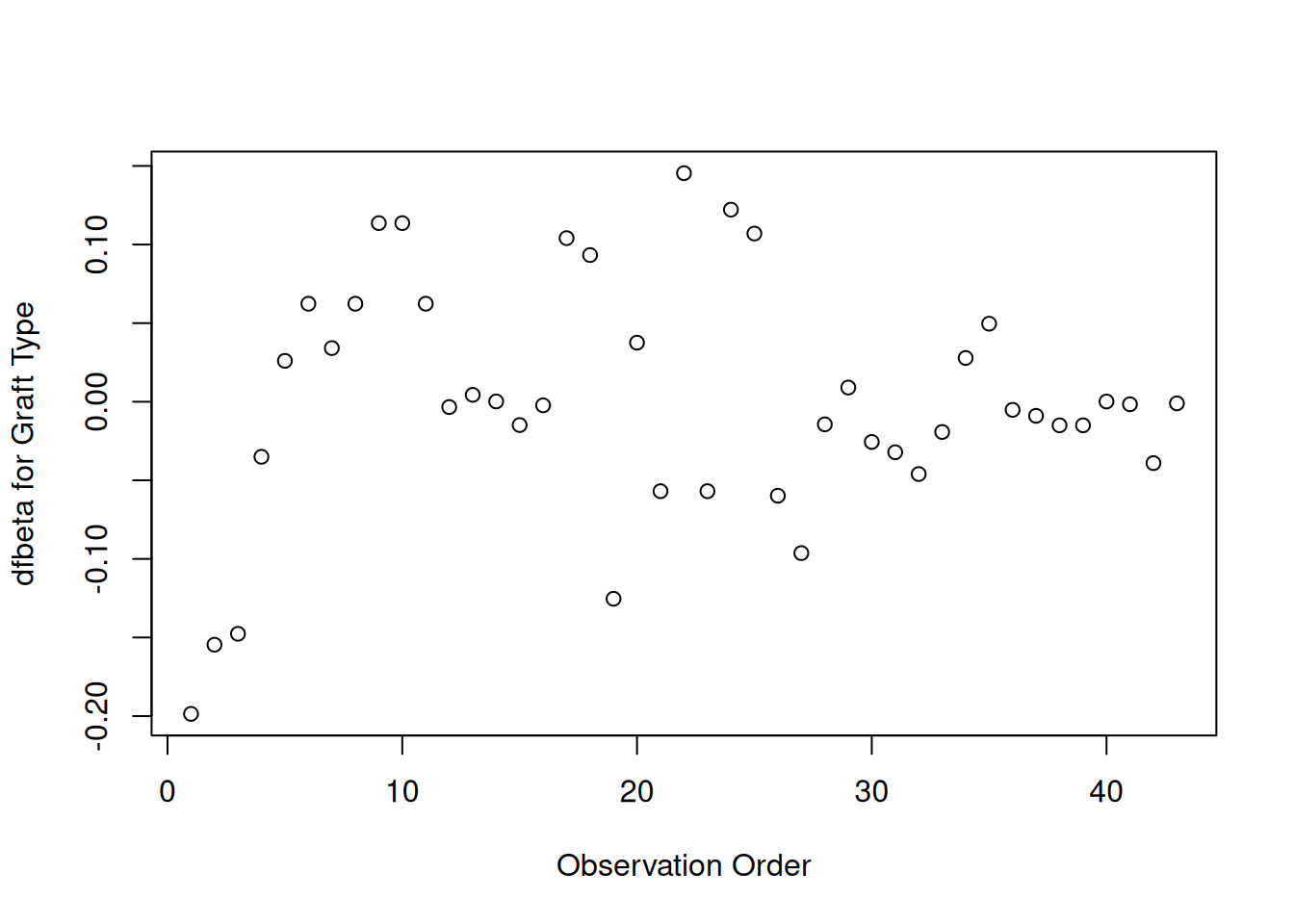
Graft type
[R code]
plot(hodg.dfb[,1],xlab="Observation Order",ylab="dfbeta for Graft Type")
dfbeta Values by Observation Order for Graft Type
The smallest dfbeta for graft type is observation 1.
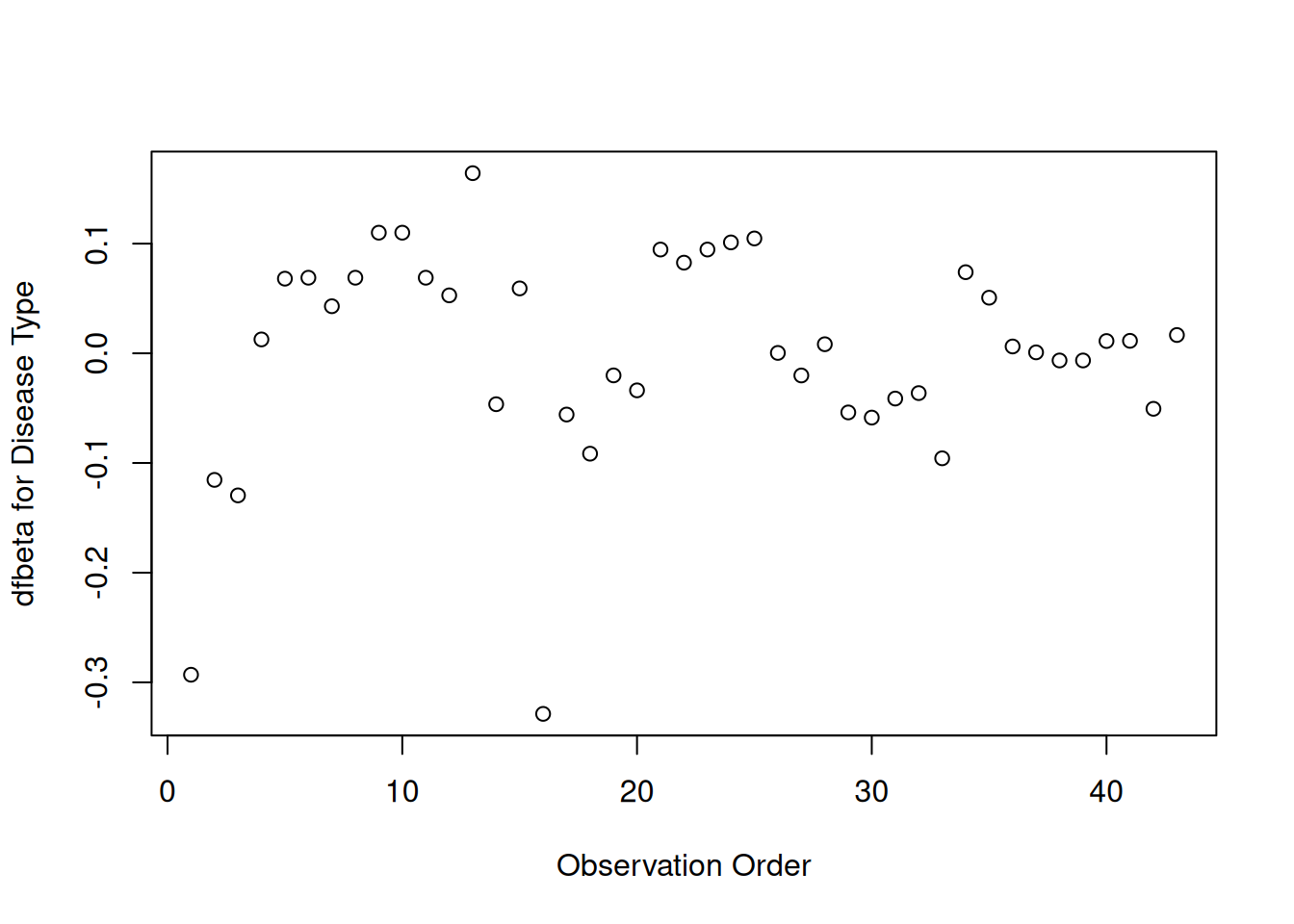
Disease type
[R code]
plot(hodg.dfb[,2],xlab="Observation Order",ylab="dfbeta for Disease Type")
dfbeta Values by Observation Order for Disease Type
The smallest two dfbeta values for disease type are observations 1 and 16.
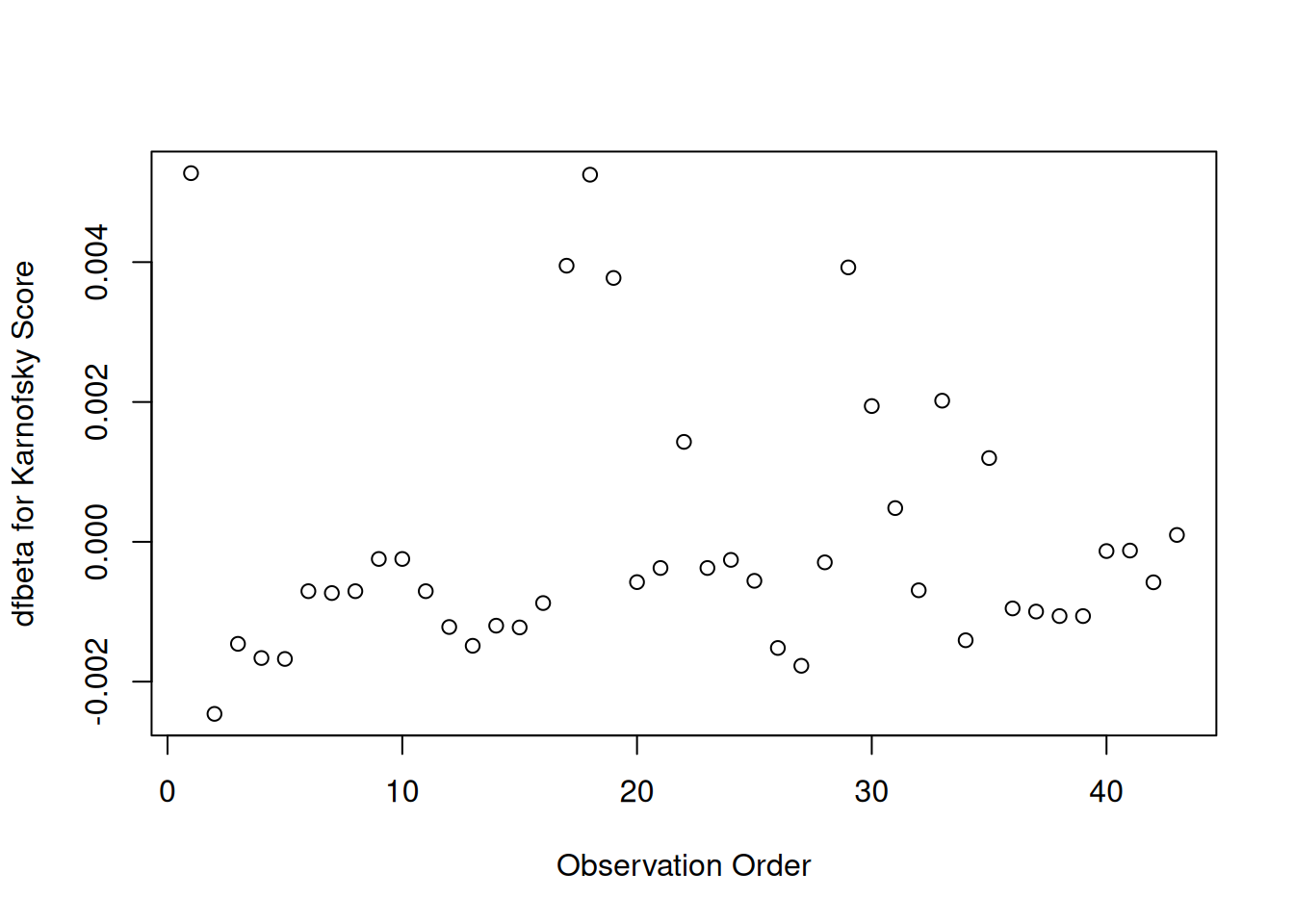
Karnofsky score
[R code]
plot(hodg.dfb[,3],xlab="Observation Order",ylab="dfbeta for Karnofsky Score")
dfbeta Values by Observation Order for Karnofsky Score
The two highest dfbeta values for score are observations 1 and 18. The next three are observations 17, 29, and 19. The smallest value is observation 2.
Waiting time (dichotomized)
[R code]
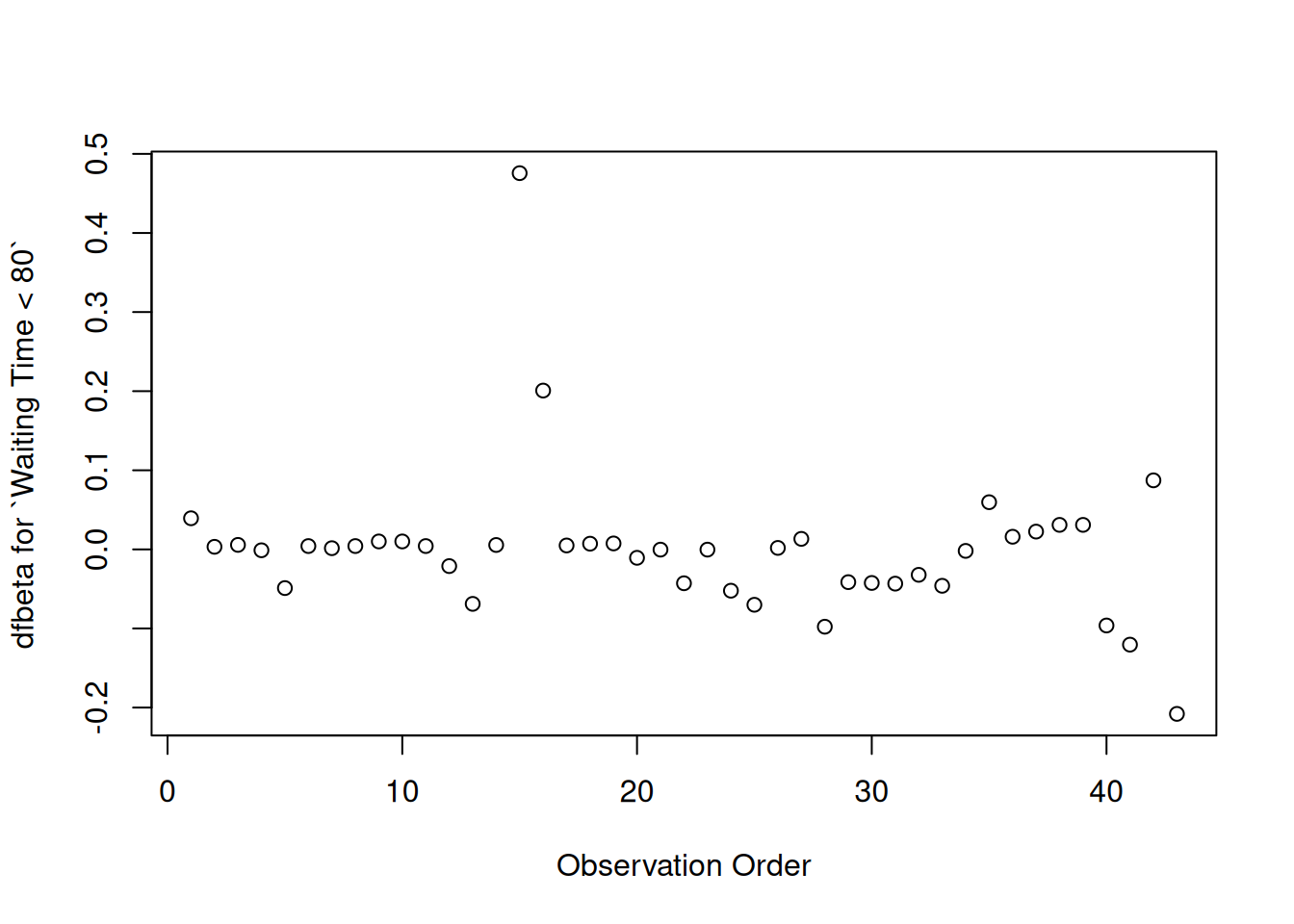
plot( hodg.dfb[,4],xlab="Observation Order",ylab="dfbeta for `Waiting Time < 80`")
dfbeta Values by Observation Order for Waiting Time (dichotomized)
The two large values of dfbeta for dichotomized waiting time are observations 15 and 16. This may have to do with the discretization of waiting time.
Interaction: graft type and disease type
[R code]
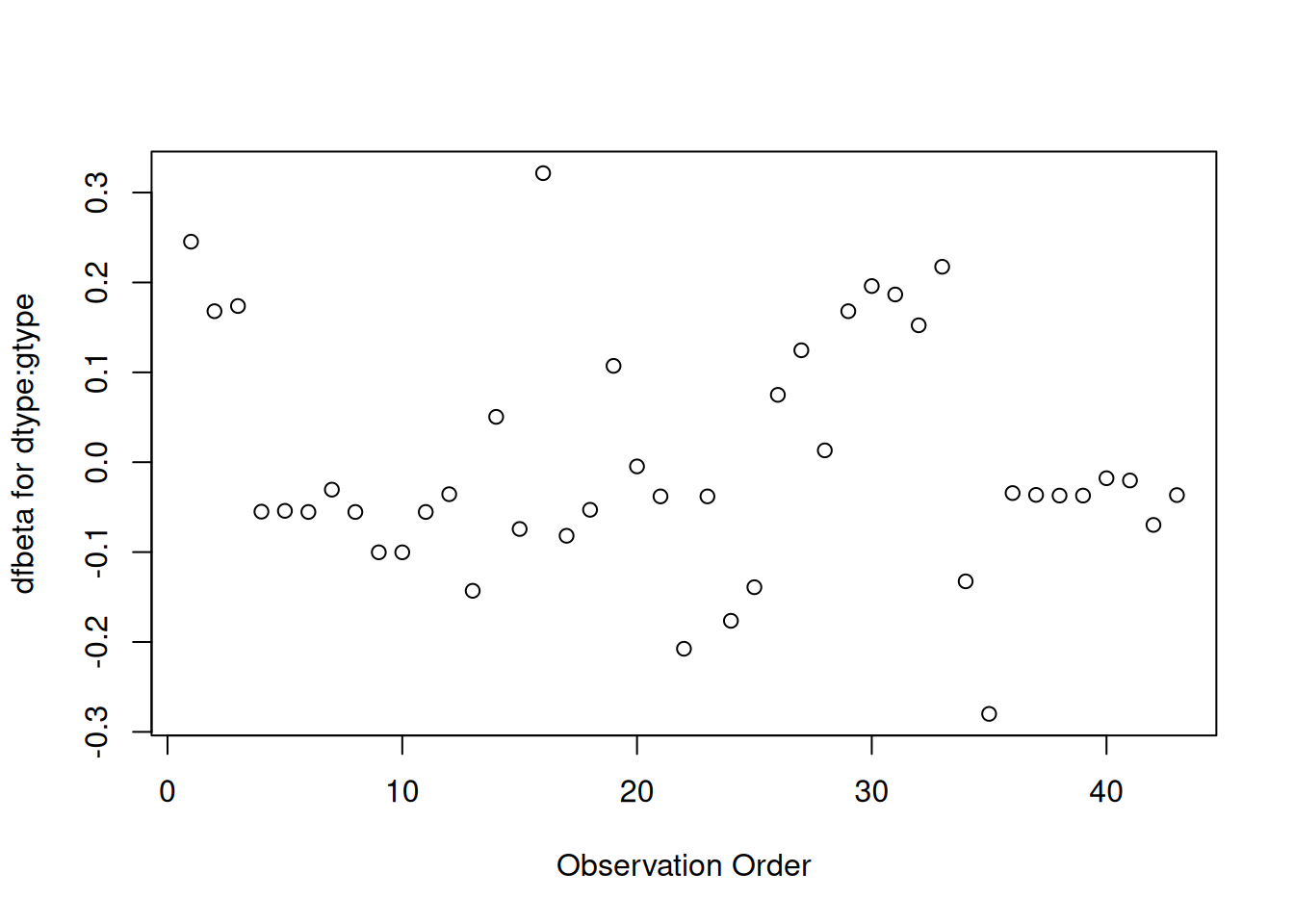
plot(hodg.dfb[,5],xlab="Observation Order",ylab="dfbeta for dtype:gtype")
dfbeta Values by Observation Order for dtype:gtype
The two largest values are observations 1 and 16. The smallest value is observation 35.
Table 2: Observations to Examine by Residuals and Influence
Diagnostic
Observations to Examine
Martingale Residuals
1, 29, 18
Deviance Residuals
1, 29
Graft Type Influence
1
Disease Type Influence
1, 16
Karnofsky Score Influence
1, 18 (17, 29, 19)
Waiting Time Influence
15, 16
Graft by Disease Influence
1, 16, 35
The most important observations to examine seem to be 1, 15, 16, 18, and 29.
[R code]
with(hodg,summary(time[delta==1]))#> Min. 1st Qu. Median Mean 3rd Qu. Max. #> 2.0 41.2 62.5 97.6 83.2 524.0
[R code]
with(hodg,summary(wtime))#> Min. 1st Qu. Median Mean 3rd Qu. Max. #> 5.0 16.0 24.0 37.7 55.5 171.0
[R code]
with(hodg,summary(score))#> Min. 1st Qu. Median Mean 3rd Qu. Max. #> 20.0 60.0 80.0 76.3 90.0 100.0
hodg2[c(1,15,16,18,29),] |>select(gtype, dtype, time, delta, score, wtime) |>mutate(comment =c("early death, good score, low risk","high risk grp, long wait, poor score","high risk grp, short wait, poor score","early death, good score, med risk grp","early death, good score, med risk grp" ))
Action Items
Unusual points may need checking, particularly if the data are not completely cleaned. In this case, observations 15 and 16 may show some trouble with the dichotomization of waiting time, but it still may be useful.
The two largest residuals seem to be due to unexpectedly early deaths, but unfortunately this can occur.
If hazards don’t look proportional, then we may need to use strata, between which the base hazards are permitted to be different. For this problem, the natural strata are the two diseases, because they could need to be managed differently anyway.
A main point that we want to be sure of is the relative risk difference by disease type and graft type.
[R code]
hodg.cox2 |>predict(reference ="zero",newdata = means |>mutate(wt2 ="short", score =0), type ="lp") |>data.frame('linear predictor'= _) |>pander()
Linear Risk Predictors for Lymphoma
linear.predictor
Non-Hodgkins,Allogenic
0
Non-Hodgkins,Autologous
0.6651
Hodgkins,Allogenic
2.327
Hodgkins,Autologous
0.9256
For Non-Hodgkin’s, the allogenic graft is better. For Hodgkin’s, the autologous graft is much better.
6.7 Stratified survival models
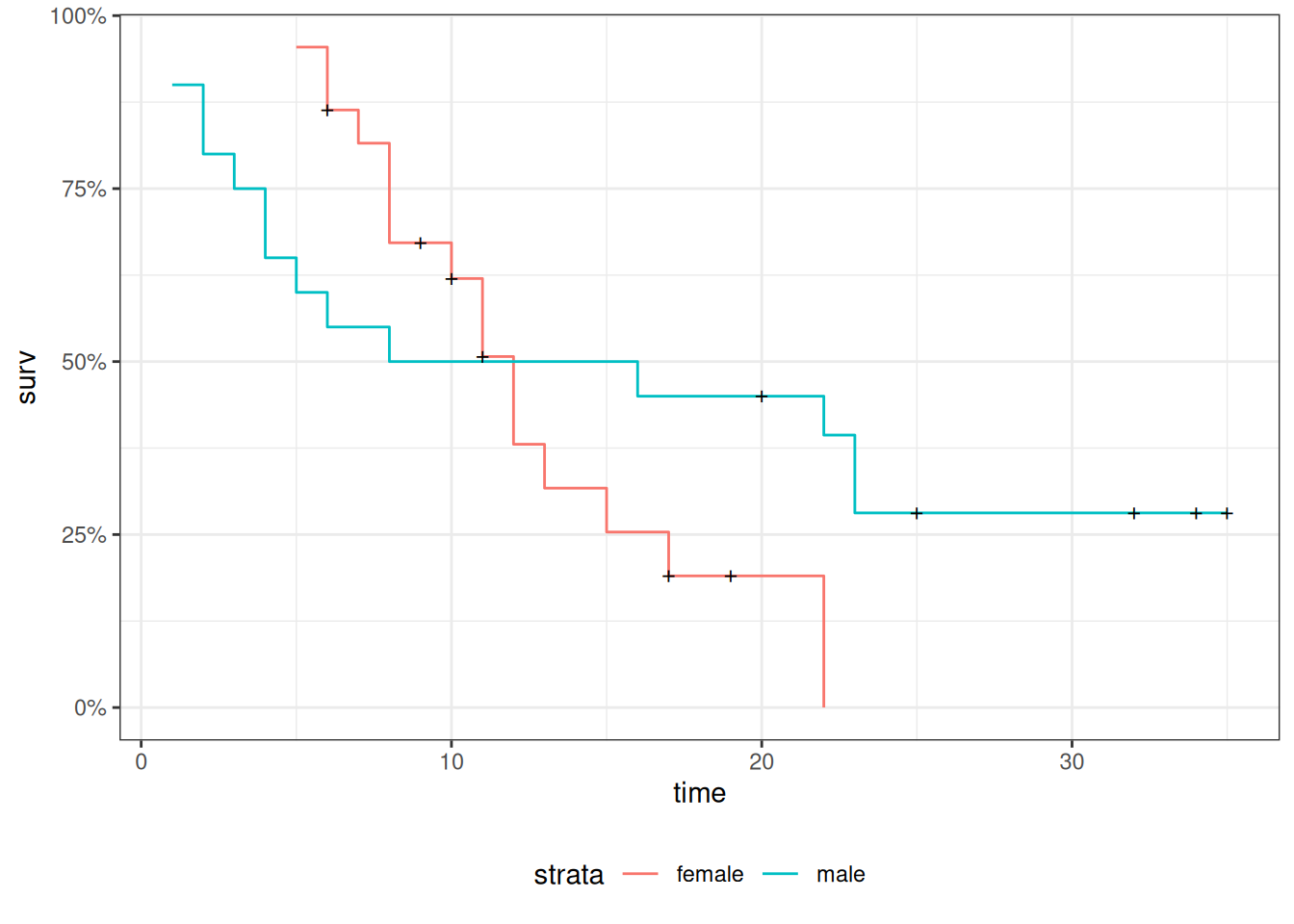
Revisiting the leukemia dataset (anderson)
We will analyze remission survival times on 42 leukemia patients, half on new treatment, half on standard treatment.
This is the same data as the drug6mp data from KMsurv, but with two other variables and without the pairing. This version comes from Kleinbaum and Klein (2012) (e.g., p281):
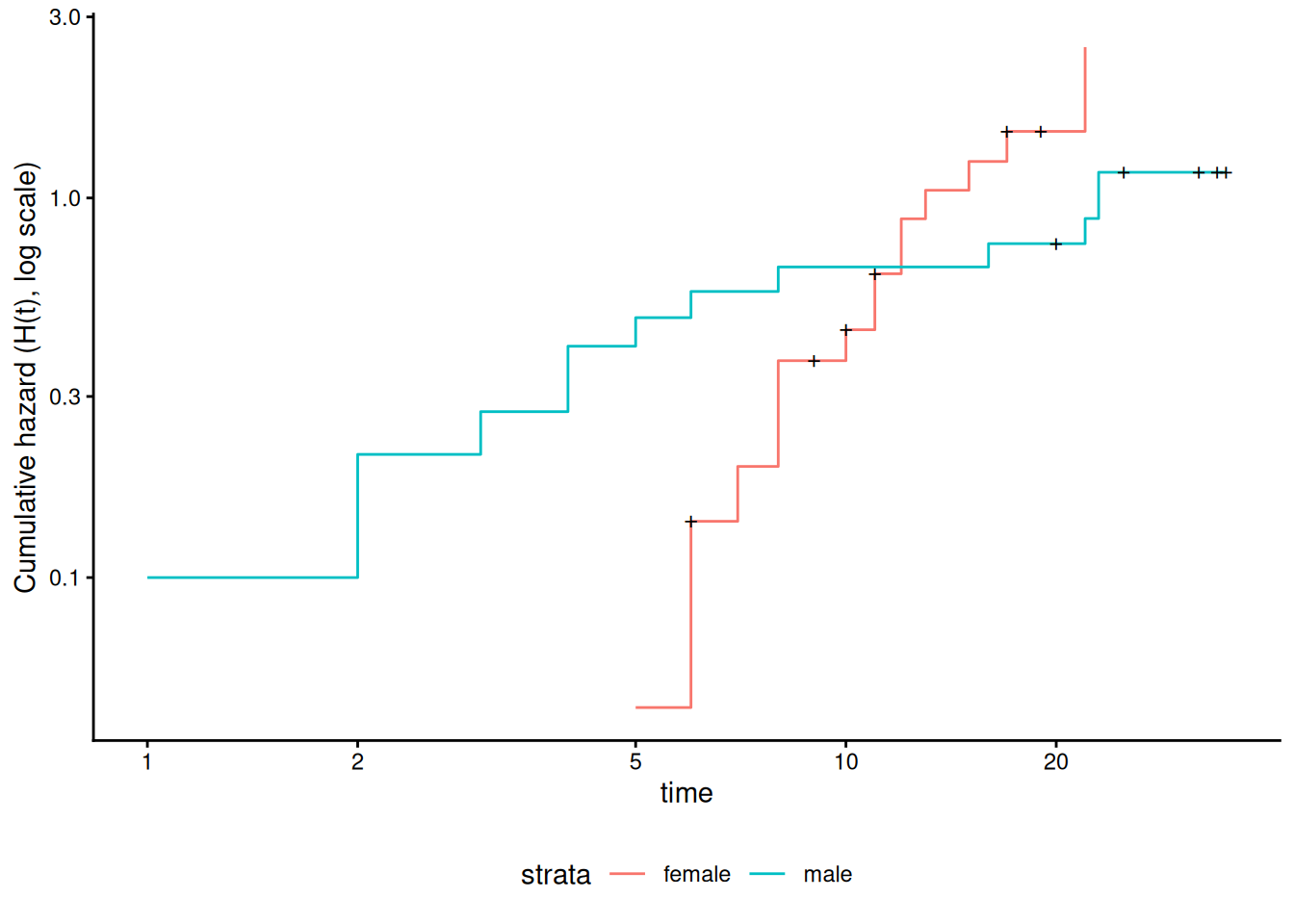
Figure 12: Cumulative hazard (cloglog scale) for anderson data
This can be fixed by using strata or possibly by other model alterations.
The Stratified Cox Model
In a stratified Cox model, each stratum, defined by one or more factors, has its own base survival function \({\lambda}_0(t)\).
But the coefficients for each variable not used in the strata definitions are assumed to be the same across strata.
To check if this assumption is reasonable one can include interactions with strata and see if they are significant (this may generate a warning and NA lines but these can be ignored).
Since the sex variable shows possible non-proportionality, we try stratifying on sex.
We don’t have enough evidence to tell the difference between these two models.
Conclusions
We chose to use a stratified model because of the apparent non-proportionality of the hazard for the sex variable.
When we fit interactions with the strata variable, we did not get an improved model (via the likelihood ratio test).
So we use the stratifed model with coefficients that are the same across strata.
Another Modeling Approach
We used an additive model without interactions and saw that we might need to stratify by sex.
Instead, we could try to improve the model’s functional form - maybe the interaction of treatment and sex is real, and after fitting that we might not need separate hazard functions.
This dataset comes from the Copelan et al. (1991) study of allogenic bone marrow transplant therapy for acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL).
Outcomes (endpoints)
The main endpoint is disease-free survival (t2 and d3) for the three risk groups, “ALL”, “AML Low Risk”, and “AML High Risk”.
Possible intermediate events
graft vs. host disease (GVHD), an immunological rejection response to the transplant (bad)
acute (AGVHD)
chronic (CGVHD)
platelet recovery, a return of platelet count to normal levels (good)
One or the other, both in either order, or neither may occur.
Covariates
We are interested in possibly using the covariates z1-z10 to adjust for other factors.
In addition, the time-varying covariates for acute GVHD, chronic GVHD, and platelet recovery may be useful.
Preprocessing
We reformat the data before analysis:
[R code]
# reformat the data:bmt1 = bmt |>as_tibble() |>mutate(id =1:n(), # will be used to connect multiple records for the same individualgroup = group |>case_match(1~"ALL",2~"Low Risk AML",3~"High Risk AML") |>factor(levels =c("ALL", "Low Risk AML", "High Risk AML")),`patient age`= z1,`donor age`= z2,`patient sex`= z3 |>case_match(0~"Female",1~"Male"),`donor sex`= z4 |>case_match(0~"Female",1~"Male"),`Patient CMV Status`= z5 |>case_match(0~"CMV Negative",1~"CMV Positive"),`Donor CMV Status`= z6 |>case_match(0~"CMV Negative",1~"CMV Positive"),`Waiting Time to Transplant`= z7,FAB = z8 |>case_match(1~"Grade 4 Or 5 (AML only)",0~"Other") |>factor() |>relevel(ref ="Other"),hospital = z9 |># `z9` is hospitalcase_match(1~"Ohio State University",2~"Alferd",3~"St. Vincent",4~"Hahnemann") |>factor() |>relevel(ref ="Ohio State University"),MTX = (z10 ==1) # a prophylatic treatment for GVHD ) |>select(-(z1:z10)) # don't need these anymorebmt1 |>select(group, id:MTX) |>print(n =10)#> # A tibble: 137 × 12#> group id `patient age` `donor age` `patient sex` `donor sex`#> <fct> <int> <int> <int> <chr> <chr> #> 1 ALL 1 26 33 Male Female #> 2 ALL 2 21 37 Male Male #> 3 ALL 3 26 35 Male Male #> 4 ALL 4 17 21 Female Male #> 5 ALL 5 32 36 Male Male #> 6 ALL 6 22 31 Male Male #> 7 ALL 7 20 17 Male Female #> 8 ALL 8 22 24 Male Female #> 9 ALL 9 18 21 Female Male #> 10 ALL 10 24 40 Male Male #> # ℹ 127 more rows#> # ℹ 6 more variables: `Patient CMV Status` <chr>, `Donor CMV Status` <chr>,#> # `Waiting Time to Transplant` <int>, FAB <fct>, hospital <fct>, MTX <lgl>
Time-Dependent Covariates
A time-dependent covariate (“TDC”) is a covariate whose value changes during the course of the study.
For variables like age that change in a linear manner with time, we can just use the value at the start.
But it may be plausible that when and if GVHD occurs, the risk of relapse or death increases, and when and if platelet recovery occurs, the risk decreases.
Analysis in R
We form a variable precovery which is = 0 before platelet recovery and is = 1 after platelet recovery, if it occurs.
For each subject where platelet recovery occurs, we set up multiple records (lines in the data frame); for example one from t = 0 to the time of platelet recovery, and one from that time to relapse, recovery, or death.
We do the same for acute GVHD and chronic GVHD.
For each record, the covariates are constant.
[R code]
bmt2 = bmt1 |>#set up new long-format data set:tmerge(bmt1, id = id, tstop = t2) |># the following three steps can be in any order, # and will still produce the same result:#add aghvd as tdc:tmerge(bmt1, id = id, agvhd =tdc(ta)) |>#add cghvd as tdc:tmerge(bmt1, id = id, cgvhd =tdc(tc)) |>#add platelet recovery as tdc:tmerge(bmt1, id = id, precovery =tdc(tp)) bmt2 = bmt2 |>as_tibble() |>mutate(status =as.numeric((tstop == t2) & d3))# status only = 1 if at end of t2 and not censored
Let’s see how we’ve rearranged the first row of the data:
[R code]
bmt1 |> dplyr::filter(id ==1) |> dplyr::select(id, t1, d1, t2, d2, d3, ta, da, tc, dc, tp, dp)
The event times for this individual are:
t = 0 time of transplant
tp = 13 platelet recovery
ta = 67 acute GVHD onset
tc = 121 chronic GVHD onset
t2 = 2081 end of study, patient not relapsed or dead
After converting the data to long-format, we have:
Note that status could have been 1 on the last row, indicating that relapse or death occurred; since it is false, the participant must have exited the study without experiencing relapse or death (i.e., they were censored).
Event sequences
Let:
A = acute GVHD
C = chronic GVHD
P = platelet recovery
Each of the eight possible combinations of A or not-A, with C or not-C, with P or not-P occurs in this data set.
A always occurs before C, and P always occurs before C, if both occur.
Thus there are ten event sequences in the data set: None, A, C, P, AC, AP, PA, PC, APC, and PAC.
In general, there could be as many as \(1+3+(3)(2)+6=16\) sequences, but our domain knowledge tells us that some are missing: CA, CP, CAP, CPA, PCA, PC, PAC
Different subjects could have 1, 2, 3, or 4 intervals, depending on which of acute GVHD, chronic GVHD, and/or platelet recovery occurred.
The final interval for any subject has status = 1 if the subject relapsed or died at that time; otherwise status = 0.
Any earlier intervals have status = 0.
Even though there might be multiple lines per ID in the dataset, there is never more than one event, so no alterations need be made in the estimation procedures or in the interpretation of the output.
The function tmerge in the survival package eases the process of constructing the new long-format dataset.
Neither acute GVHD (agvhd) nor chronic GVHD (cgvhd) has a statistically significant effect here, nor are they significant in models with the other one removed.
Sometimes an appropriate analysis requires consideration of recurrent events.
A patient with arthritis may have more than one flareup. The same is true of many recurring-remitting diseases.
In this case, we have more than one line in the data frame, but each line may have an event.
We have to use a “robust” variance estimator to account for correlation of time-to-events within a patient.
Bladder Cancer Data Set
The bladder cancer dataset from Kleinbaum and Klein (2012) contains recurrent event outcome information for eighty-six cancer patients followed for the recurrence of bladder cancer tumor after transurethral surgical excision (Byar and Green 1980). The exposure of interest is the effect of the drug treatment of thiotepa. Control variables are the initial number and initial size of tumors. The data layout is suitable for a counting processes approach.
This drug is still a possible choice for some patients. Another therapeutic choice is Bacillus Calmette-Guerin (BCG), a live bacterium related to cow tuberculosis.
Data dictionary
Variables in the bladder dataset
Variable
Definition
id
Patient unique ID
status
for each time interval: 1 = recurred, 0 = censored
interval
1 = first recurrence, etc.
intime
`tstop - tstart (all times in months)
tstart
start of interval
tstop
end of interval
tx
treatment code, 1 = thiotepa
num
number of initial tumors
size
size of initial tumors (cm)
There are 85 patients and 190 lines in the dataset, meaning that many patients have more than one line.
Patient 1 with 0 observation time was removed.
Of the 85 patients, 47 had at least one recurrence and 38 had none.
18 patients had exactly one recurrence.
There were up to 4 recurrences in a patient.
Of the 190 intervals, 112 terminated with a recurrence and 78 were censored.
Different intervals for the same patient are correlated.
Is the effective sample size 47 or 112? This might narrow confidence intervals by as much as a factor of \(\sqrt{112/47}=1.54\)
What happens if I have 5 treatment and 5 control values and want to do a t-test and I then duplicate the 10 values as if the sample size was 20? This falsely narrows confidence intervals by a factor of \(\sqrt{2}=1.41\).
bladder = bladder |>mutate(surv =Surv(time = start,time2 = stop,event = event,type ="counting"))bladder.cox1 =coxph(formula = surv~tx+num+size,data = bladder)#results with biased variance-covariance matrix:summary(bladder.cox1)#> Call:#> coxph(formula = surv ~ tx + num + size, data = bladder)#> #> n= 190, number of events= 112 #> #> coef exp(coef) se(coef) z Pr(>|z|) #> tx -0.4116 0.6626 0.1999 -2.06 0.03947 * #> num 0.1637 1.1778 0.0478 3.43 0.00061 ***#> size -0.0411 0.9598 0.0703 -0.58 0.55897 #> ---#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1#> #> exp(coef) exp(-coef) lower .95 upper .95#> tx 0.663 1.509 0.448 0.98#> num 1.178 0.849 1.073 1.29#> size 0.960 1.042 0.836 1.10#> #> Concordance= 0.624 (se = 0.032 )#> Likelihood ratio test= 14.7 on 3 df, p=0.002#> Wald test = 15.9 on 3 df, p=0.001#> Score (logrank) test = 16.2 on 3 df, p=0.001
Note
The likelihood ratio and score tests assume independence of observations within a cluster. The Wald and robust score tests do not.
adding cluster = id
If we add cluster= id to the call to coxph, the coefficient estimates don’t change, but we get an additional column in the summary() output: robust se:
[R code]
bladder.cox2 =coxph(formula = surv ~ tx + num + size,cluster = id,data = bladder)#unbiased though this reduces power:summary(bladder.cox2)#> Call:#> coxph(formula = surv ~ tx + num + size, data = bladder, cluster = id)#> #> n= 190, number of events= 112 #> #> coef exp(coef) se(coef) robust se z Pr(>|z|) #> tx -0.4116 0.6626 0.1999 0.2488 -1.65 0.0980 . #> num 0.1637 1.1778 0.0478 0.0584 2.80 0.0051 **#> size -0.0411 0.9598 0.0703 0.0742 -0.55 0.5799 #> ---#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1#> #> exp(coef) exp(-coef) lower .95 upper .95#> tx 0.663 1.509 0.407 1.08#> num 1.178 0.849 1.050 1.32#> size 0.960 1.042 0.830 1.11#> #> Concordance= 0.624 (se = 0.031 )#> Likelihood ratio test= 14.7 on 3 df, p=0.002#> Wald test = 11.2 on 3 df, p=0.01#> Score (logrank) test = 16.2 on 3 df, p=0.001, Robust = 10.8 p=0.01#> #> (Note: the likelihood ratio and score tests assume independence of#> observations within a cluster, the Wald and robust score tests do not).
robust se is larger than se, and accounts for the repeated observations from the same individuals:
Copelan, Edward A, James C Biggs, James M Thompson, et al. 1991. Treatment for Acute Myelocytic Leukemia with Allogeneic Bone Marrow Transplantation Following Preparation with BuCy2. https://doi.org/10.1182/blood.V78.3.838.838.
Dobson, Annette J, and Adrian G Barnett. 2018. An Introduction to Generalized Linear Models. 4th ed. CRC press. https://doi.org/10.1201/9781315182780.
Grambsch, Patricia M, and Terry M Therneau. 1994. “Proportional Hazards Tests and Diagnostics Based on Weighted Residuals.”Biometrika 81 (3): 515–26. https://doi.org/10.1093/biomet/81.3.515.